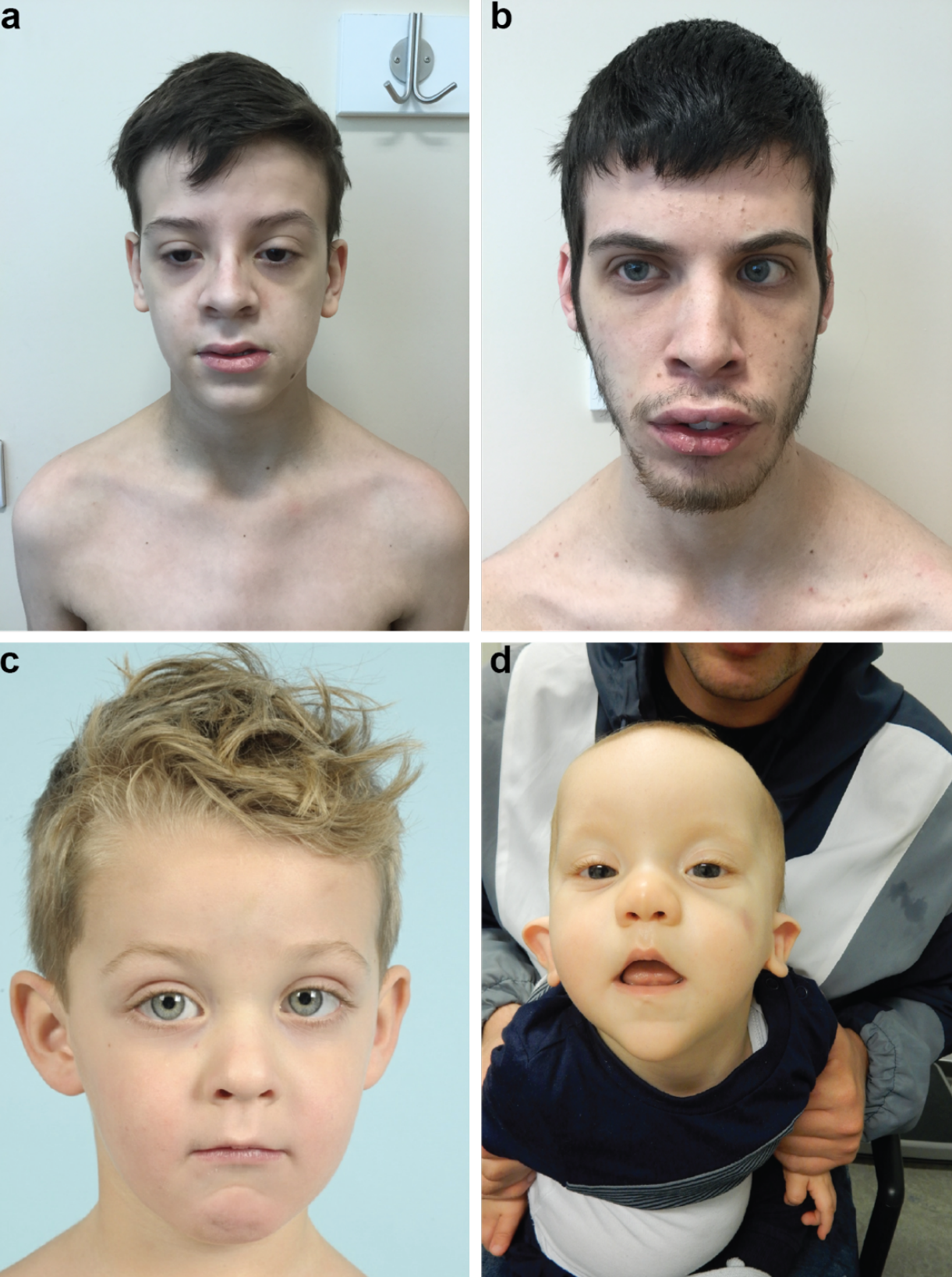
Uma síndrome rara com múltiplas alterações presentes desde o nascimento, caracterizada por altura acima da média, deficiência intelectual leve a moderada e uma aparência facial peculiar, como rosto arredondado, sobrancelhas grossas e horizontais, e olhos com abertura estreita.
Introdução
O que você precisa saber de cara
Uma síndrome rara com múltiplas alterações presentes desde o nascimento, caracterizada por altura acima da média, deficiência intelectual leve a moderada e uma aparência facial peculiar, como rosto arredondado, sobrancelhas grossas e horizontais, e olhos com abertura estreita.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 66 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisRequired for genome-wide de novo methylation and is essential for the establishment of DNA methylation patterns during development (PubMed:12138111, PubMed:16357870, PubMed:30478443). DNA methylation is coordinated with methylation of histones (PubMed:12138111, PubMed:16357870, PubMed:30478443). It modifies DNA in a non-processive manner and also methylates non-CpG sites (PubMed:12138111, PubMed:16357870, PubMed:30478443). May preferentially methylate DNA linker between 2 nucleosomal cores and i
NucleusChromosomeCytoplasm
Tatton-Brown-Rahman syndrome
An overgrowth syndrome characterized by a distinctive facial appearance, tall stature and intellectual disability. Facial gestalt is characterized by a round face, heavy horizontal eyebrows and narrow palpebral fissures. Less common features include atrial septal defects, seizures, umbilical hernia, and scoliosis.
Variantes genéticas (ClinVar)
362 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Tatton-Brown-Rahman
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Familial pneumothorax in twins with Tatton-Brown-Rahman DNMT3A overgrowth syndrome.
Spontaneous pneumothorax is a common respiratory presentation that may signal underlying genetic disease. Familial pneumothorax occurs in ~10% of primary cases, yet 75% remain genetically unclassified. We report identical twin brothers presenting with spontaneous pneumothoraces in adulthood, leading to a diagnosis of Tatton-Brown-Rahman syndrome (TBRS), a DNMT3A-related overgrowth disorder not previously associated with pneumothorax. Both individuals exhibited tall stature, mild intellectual disability, hypermobility, and cardiac abnormalities. Whole genome sequencing identified a rare de novo DNMT3A missense variant (c.1585 G > A, p.D529N) absent from population databases and predicted to be damaging. Methylation profiling confirmed genome-wide hypomethylation consistent with impaired DNMT3A function, supporting pathogenicity. No variants were found in known familial pneumothorax genes. Apical blebs observed at surgery and connective tissue features suggest a mechanistic link between TBRS and pneumothorax, analogous to other monogenic connective tissue disorders. This case expands the phenotypic spectrum of TBRS and highlights the importance of genetic evaluation in familial pneumothorax. Diagnosis enables personalised care, including surveillance for extrapulmonary complications such as aortic root dilatation and haematological malignancy. Our findings suggest that TBRS should be considered in patients presenting with pneumothorax, tall stature, and neurodevelopmental features. Further cases are needed to confirm this association and refine clinical management strategies.
Expanding the Malignancy Spectrum of Tatton-Brown-Rahman Syndrome: A Case of Hodgkin Lymphoma.
DNMT3A R882C variant in a patient with a presumed pineal gland tumor, highlighting potential tumor susceptibility in Tatton-Brown-Rahman syndrome.
Tatton-Brown-Rahman syndrome (TBRS) is a rare overgrowth disorder caused by germline alterations in DNMT3A, a gene essential for DNA methylation and epigenetic regulation. Affected individuals typically present with tall stature, intellectual disability, and neurodevelopmental disorders, and they also demonstrate an increased susceptibility to neoplastic disease. Reported tumors include hematologic malignancies, with acute myeloid leukemia being the most notable, as well as solid tumors such as neuroblastoma, medulloblastoma, benign glioma, and pituitary adenoma. Despite this expanding tumor spectrum, pineal gland tumors have not previously been described in association with TBRS. Here, we present the first known case of a presumed pineal gland tumor in a patient with TBRS. Although the lesion lacks histopathologic confirmation and a sporadic occurrence cannot be excluded, this observation raises the possibility that germline DNMT3A alterations may predispose to a broader range of tumors than previously recognized. This report underscores the need for continued documentation of unusual tumor presentations in TBRS to further elucidate the biology of DNMT3A-related tumorigenesis.
Epileptic encephalopathy with spike-and-wave activation in sleep associated with Tatton-Brown-Rahman syndrome responsive to highly purified cannabidiol.
Tatton-Brown-Rahman Syndrome Due to a Novel DNMT3A Variant Presenting With Autism, Attention-Deficit/Hyperactivity Disorder (ADHD), and Regression: A Saudi Case Report.
Tatton-Brown-Rahman syndrome (TBRS) is a rare overgrowth/intellectual disability disorder caused by DNMT3A variants. It is characterized by tall stature, macrocephaly, and intellectual disability, with an expanding neuropsychiatric spectrum that includes autism spectrum disorder (ASD) and behavioral disturbances. We describe a young adult Saudi male with overgrowth, macrocephaly, coarse facial features, and global developmental delay. Cognitive testing confirmed mild intellectual disability with relative verbal strengths. He was diagnosed with ASD and attention-deficit/hyperactivity disorder (ADHD) and treated with methylphenidate in childhood. In early adulthood, the patient exhibited regression characterized by speech loss, decline in self-care, incontinence, and psychotic-like symptoms, alongside gastrointestinal disease. Genetic testing revealed a novel heterozygous DNMT3A missense variant (c.923G>T, p.Gly308Val) in the PWWP domain, consistent with TBRS. This case illustrates the broadened phenotype of TBRS, including ASD, ADHD, and regression, paralleling prior reports of cognitive unevenness and behavioral vulnerability. Additional findings of eczema, elevated IgE, and anemia suggest possible underrecognized systemic involvement. This case emphasizes the importance of multidisciplinary evaluation and lifelong neuropsychiatric follow-up in TBRS. As the first reported case from Saudi Arabia to our knowledge, it broadens the clinical and geographic spectrum of the disorder and highlights the association between DNMT3A variants, intellectual disability, and ASD.
Publicações recentes
Epileptic encephalopathy with spike-and-wave activation in sleep associated with Tatton-Brown-Rahman syndrome responsive to highly purified cannabidiol.
DNMT3A R882C variant in a patient with a presumed pineal gland tumor, highlighting potential tumor susceptibility in Tatton-Brown-Rahman syndrome.
Familial pneumothorax in twins with Tatton-Brown-Rahman DNMT3A overgrowth syndrome.
Expanding the Malignancy Spectrum of Tatton-Brown-Rahman Syndrome: A Case of Hodgkin Lymphoma.
Tatton-Brown-Rahman Syndrome Due to a Novel DNMT3A Variant Presenting With Autism, Attention-Deficit/Hyperactivity Disorder (ADHD), and Regression: A Saudi Case Report.
📚 EuropePMC38 artigos no totalmostrando 57
Epileptic encephalopathy with spike-and-wave activation in sleep associated with Tatton-Brown-Rahman syndrome responsive to highly purified cannabidiol.
Epileptic disorders : international epilepsy journal with videotapeDNMT3A R882C variant in a patient with a presumed pineal gland tumor, highlighting potential tumor susceptibility in Tatton-Brown-Rahman syndrome.
Cancer geneticsFamilial pneumothorax in twins with Tatton-Brown-Rahman DNMT3A overgrowth syndrome.
European journal of human genetics : EJHGExpanding the Malignancy Spectrum of Tatton-Brown-Rahman Syndrome: A Case of Hodgkin Lymphoma.
Pediatric blood & cancerTatton-Brown-Rahman Syndrome Due to a Novel DNMT3A Variant Presenting With Autism, Attention-Deficit/Hyperactivity Disorder (ADHD), and Regression: A Saudi Case Report.
CureusTatton-Brown-Rahman-Syndrome-associated DNMT3A mutations de-repress cortical interneuron differentiation to disrupt neuronal network function.
bioRxiv : the preprint server for biologyOvergrowth-intellectual disability disorders: progress in biology, patient advocacy and innovative therapies.
Disease models & mechanismsStability and DNA Methyltransferase Activity of DNMT3A are Maintained by Ubiquitin-Specific Peptidase 11 (USP11) and Sumoylation Countering Degradation.
bioRxiv : the preprint server for biologyFrom Serendipity to Scalability in Rare Disease Patient Collaborations.
Missouri medicineCase Report: A case of Tatton-Brown-Rahman syndrome featuring mitral annular disjunction and mitral valve prolapse due to a novel mutation site in the DNMT3A gene.
Frontiers in cardiovascular medicineTatton-Brown-Rahman syndrome: A new multiple endocrine neoplasia syndrome with intellectual disability?
Annales d'endocrinologieUltra-rare monogenic disorders frequently detected among sex chromosome aneuploidy patients with atypical findings.
Journal of human geneticsDNMT3A-related overgrowth syndrome presenting with immune thrombocytopenic purpura.
Current research in translational medicineComparison of Genetic, Auditory Features, and Systemic Clinical Phenotype in 14 Families with Syndromic Hearing Loss.
The application of clinical geneticsArterial aneurysm and dissection: toward the evolving phenotype of Tatton-Brown-Rahman syndrome.
Journal of medical geneticsExpanding the genetic and clinical spectrum of Tatton-Brown-Rahman syndrome in a series of 24 French patients.
Journal of medical geneticsAortic disease and cardiomyopathy in patients with a novel DNMT3A gene variant causing Tatton-Brown-Rahman syndrome.
Clinical epigeneticsEpilepsy and overgrowth-intellectual disability syndromes: a patient organization perspective on collaborating to accelerate pathways to treatment.
Therapeutic advances in rare diseaseClinical Case of Mild Tatton-Brown-Rahman Syndrome Caused by a Nonsense Variant in DNMT3A Gene.
Clinics and practiceSkeletal abnormalities in mice with Dnmt3a missense mutations.
BoneAn adult patient with Tatton-Brown-Rahman syndrome caused by a novel DNMT3A variant and axonal polyneuropathy.
American journal of medical genetics. Part AThe Role of Clonal Hematopoiesis of Indeterminant Potential and DNA (Cytosine-5)-Methyltransferase Dysregulation in Pulmonary Arterial Hypertension and Other Cardiovascular Diseases.
CellsTatton-Brown-Rahman syndrome: Novel pathogenic variants and new neuroimaging findings.
American journal of medical genetics. Part AMethylation signatures in clinically variable syndromic disorders: a familial DNMT3A variant in two adults with Tatton-Brown-Rahman syndrome.
European journal of human genetics : EJHGEpigenetic Causes of Overgrowth Syndromes.
The Journal of clinical endocrinology and metabolismA novel pathogenic variant of DNMT3A associated with craniosynostosis: a case report of Heyn-Sproul-Jackson syndrome.
Frontiers in pediatricsSensory processing in Sotos syndrome and Tatton-Brown-Rahman Syndrome.
Journal of psychopathology and clinical scienceExpanding the phenotype of DNMT3A as a cause a congenital myopathy with rhabdomyolysis.
Neuromuscular disorders : NMDTreatment of Primary Hyperparathyroidism in the Setting of Tatton-Brown-Rahman Syndrome.
The American surgeonCase Report: The success of face analysis technology in extremely rare genetic diseases in Korea: Tatton-Brown-Rahman syndrome and Say-Barber -Biesecker-Young-Simpson variant of ohdo syndrome.
Frontiers in genetics[Tatton-Brown-Rahman Syndrome: Case report and DNMT3A variant not previously reported associated to the syndrome].
Andes pediatrica : revista Chilena de pediatriaConstitutive loss of DNMT3A causes morbid obesity through misregulation of adipogenesis.
eLifeThe Epigenetic Role of Vitamin C in Neurodevelopment.
International journal of molecular sciencesCase Report: Bilateral Epiphysiodesis Due to Extreme Tall Stature in a Girl With a De Novo DNMT3A Variant Associated With Tatton-Brown-Rahman Syndrome.
Frontiers in endocrinologyAortic root dilatation and dilated cardiomyopathy in an adult with Tatton-Brown-Rahman syndrome.
American journal of medical genetics. Part APerturbed hematopoiesis in individuals with germline DNMT3A overgrowth Tatton-Brown-Rahman syndrome.
HaematologicaDnmt3a deficiency in the skin causes focal, canonical DNA hypomethylation and a cellular proliferation phenotype.
Proceedings of the National Academy of Sciences of the United States of America[Tatton-Brown-Rahman syndrome associated with the DNMT3A gene: a case report and literature review].
Zhongguo dang dai er ke za zhi = Chinese journal of contemporary pediatricsBehavioral and dental management of a patient with Tatton-Brown-Rahman syndrome: Case report.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryTatton-Brown-Rahman syndrome with a novel DNMT3A mutation presented severe intellectual disability and autism spectrum disorder.
Human genome variationTatton-Brown-Rahman syndrome: Six individuals with novel features.
American journal of medical genetics. Part AFirst identified Korean family with Tatton-Brown-Rahman Syndrome caused by the novel DNMT3A variant c.118G>C p.(Glu40Gln).
Annals of pediatric endocrinology & metabolismAcromegaly in the setting of Tatton-Brown-Rahman Syndrome.
PituitaryTatton-Brown-Rahman syndrome: cognitive and behavioural phenotypes.
Developmental medicine and child neurologyFurther delineation of neuropsychiatric findings in Tatton-Brown-Rahman syndrome due to disease-causing variants in DNMT3A: seven new patients.
European journal of human genetics : EJHGPRC2 functions in development and congenital disorders.
Development (Cambridge, England)The histone mark H3K36me2 recruits DNMT3A and shapes the intergenic DNA methylation landscape.
NatureGrowth disrupting mutations in epigenetic regulatory molecules are associated with abnormalities of epigenetic aging.
Genome researchThe first case report of medulloblastoma associated with Tatton-Brown-Rahman syndrome.
American journal of medical genetics. Part AThe Tatton-Brown-Rahman Syndrome: A clinical study of 55 individuals with de novo constitutive DNMT3A variants.
Wellcome open researchCo-occurrence of a maternally inherited DNMT3A duplication and a paternally inherited pathogenic variant in EZH2 in a child with growth retardation and severe short stature: atypical Weaver syndrome or evidence of a DNMT3A dosage effect?
Cold Spring Harbor molecular case studiesThe spectrum of DNMT3A variants in Tatton-Brown-Rahman syndrome overlaps with that in hematologic malignancies.
American journal of medical genetics. Part AA case of familial transmission of the newly described DNMT3A-Overgrowth Syndrome.
American journal of medical genetics. Part AAcute myeloid leukaemia in a case with Tatton-Brown-Rahman syndrome: the peculiar DNMT3A R882 mutation.
Journal of medical geneticsPathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome.
Human mutationNovel DNMT3A germline mutations are associated with inherited Tatton-Brown-Rahman syndrome.
Clinical geneticsTatton-Brown-Rahman syndrome due to 2p23 microdeletion.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Tatton-Brown-Rahman.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Tatton-Brown-Rahman
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Familial pneumothorax in twins with Tatton-Brown-Rahman DNMT3A overgrowth syndrome.
- Expanding the Malignancy Spectrum of Tatton-Brown-Rahman Syndrome: A Case of Hodgkin Lymphoma.
- DNMT3A R882C variant in a patient with a presumed pineal gland tumor, highlighting potential tumor susceptibility in Tatton-Brown-Rahman syndrome.
- Epileptic encephalopathy with spike-and-wave activation in sleep associated with Tatton-Brown-Rahman syndrome responsive to highly purified cannabidiol.
- Tatton-Brown-Rahman Syndrome Due to a Novel DNMT3A Variant Presenting With Autism, Attention-Deficit/Hyperactivity Disorder (ADHD), and Regression: A Saudi Case Report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:404443(Orphanet)
- OMIM OMIM:615879(OMIM)
- MONDO:0014382(MONDO)
- GARD:17674(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q53160608(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Tatton-Brown-Rahman
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata