A artrogripose múltipla congênita (AMC) é um grupo de doenças caracterizadas por contraturas congênitas dos membros. Manifesta-se como limitação do movimento de múltiplas articulações dos membros ao nascimento, que geralmente não é progressiva e pode incluir fraqueza muscular e fibrose. AMC está sempre associada à diminuição do movimento fetal intrauterino que leva secundariamente às contraturas.
Introdução
O que você precisa saber de cara
A artrogripose múltipla congênita (AMC) é um grupo de doenças caracterizadas por contraturas congênitas dos membros. Manifesta-se como limitação do movimento de múltiplas articulações dos membros ao nascimento, que geralmente não é progressiva e pode incluir fraqueza muscular e fibrose. AMC está sempre associada à diminuição do movimento fetal intrauterino que leva secundariamente às contraturas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 213 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 504 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
30 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable, X-linked recessive.
Curadoria gene-doença
fontes oficiaisProcessive microtubule plus-end directed motor protein involved in neuronal axon guidance. Is recruited by KANK1 to cortical microtubule stabilizing complexes (CMSCs) at focal adhesions (FAs) rims where it promotes microtubule capture and stability. Controls microtubule polymerization rate at axonal growth cones and suppresses microtubule growth without inducing microtubule disassembly once it reaches the cell cortex
Cytoplasm, cytoskeletonCytoplasm, cell cortexCell projection, axonCell projection, dendriteCell projection, growth cone
Fibrosis of extraocular muscles, congenital, 1
A congenital ocular motility disorder marked by restrictive ophthalmoplegia affecting extraocular muscles innervated by the oculomotor and/or trochlear nerves. It is clinically characterized by anchoring of the eyes in downward gaze, ptosis, and backward tilt of the head. Patients affected by congenital fibrosis of extraocular muscles type 1 show an absence of the superior division of the oculomotor nerve (cranial nerve III) and corresponding oculomotor subnuclei.
Thick filament-associated protein located in the crossbridge region of vertebrate striated muscle a bands. Slow skeletal protein that binds to both myosin and actin (PubMed:31025394, PubMed:31264822). In vitro, binds to native thin filaments and modifies the activity of actin-activated myosin ATPase. May modulate muscle contraction or may play a more structural role
Arthrogryposis, distal, 1B
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. Distal arthrogryposis type 1 is characterized largely by camptodactyly and clubfoot. Hypoplasia and/or absence of some interphalangeal creases is common. The shoulders and hips are less frequently affected.
Protein with chaperone functions important for the control of protein folding, processing, stability and localization as well as for the reduction of misfolded protein aggregates. Involved in the regulation of synaptic vesicle recycling, controls STON2 protein stability in collaboration with the COP9 signalosome complex (CSN). In the nucleus, may link the cytoskeleton with the nuclear envelope, this mechanism seems to be crucial for the control of nuclear polarity, cell movement and, specificall
Endoplasmic reticulum lumenNucleus membraneCell projection, growth coneCytoplasmic vesicle membraneCytoplasmic vesicle, secretory vesicleCytoplasmic vesicle, secretory vesicle, synaptic vesicleCytoplasm, cytoskeleton
Dystonia 1, torsion, autosomal dominant
A primary torsion dystonia, and the most common and severe form. Dystonia is defined by the presence of sustained involuntary muscle contractions, often leading to abnormal postures. Dystonia type 1 is characterized by involuntary, repetitive, sustained muscle contractions or postures involving one or more sites of the body, in the absence of other neurological symptoms. Typically, symptoms develop first in an arm or leg in middle to late childhood and progress in approximately 30% of patients to other body regions (generalized dystonia) within about five years. 'Torsion' refers to the twisting nature of body movements observed in DYT1, often affecting the trunk. Distribution and severity of symptoms vary widely between affected individuals, ranging from mild focal dystonia to severe generalized dystonia, even within families.
Probable muscle-intrinsic activator of MUSK that plays an essential role in neuromuscular synaptogenesis. Acts in aneural activation of MUSK and subsequent acetylcholine receptor (AchR) clustering in myotubes. Induces autophosphorylation of MUSK
Cell membraneSynapse
Myasthenic syndrome, congenital, 10
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS10 is an autosomal recessive, post-synaptic form characterized by a typical 'limb girdle' pattern of muscle weakness with small, simplified neuromuscular junctions but normal acetylcholine receptor and acetylcholinesterase function.
Ligand for NRCAM and NFASC/neurofascin that plays a role in the formation and maintenance of the nodes of Ranvier on myelinated axons. Mediates interaction between Schwann cell microvilli and axons via its interactions with NRCAM and NFASC. Nodes of Ranvier contain clustered sodium channels that are crucial for the saltatory propagation of action potentials along myelinated axons. During development, nodes of Ranvier are formed by the fusion of two heminodes. Required for normal clustering of so
Cell membraneCell projection, axonSecretedSecreted, extracellular space, extracellular matrix
Lethal congenital contracture syndrome 11
A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy and congenital non-progressive joint contractures. The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth.
Multi-isomeric modular protein which forms a linking network between organelles and the actin cytoskeleton to maintain the subcellular spatial organization. As a component of the LINC (LInker of Nucleoskeleton and Cytoskeleton) complex involved in the connection between the nuclear lamina and the cytoskeleton. The nucleocytoplasmic interactions established by the LINC complex play an important role in the transmission of mechanical forces across the nuclear envelope and in nuclear movement and p
Nucleus outer membraneNucleusNucleus envelopeCytoplasm, cytoskeletonCytoplasm, myofibril, sarcomereGolgi apparatus
Spinocerebellar ataxia, autosomal recessive, 8
A form of spinocerebellar ataxia, a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCAR8 is an autosomal recessive form.
Microtubule motor protein that binds to microtubules with high affinity through each tubulin heterodimer and has an ATPase activity (By similarity). Plays a role in many processes like cell division, cytokinesis and also in cell proliferation and apoptosis (PubMed:16648480, PubMed:24784001). During cytokinesis, targets to central spindle and midbody through its interaction with PRC1 and CIT respectively (PubMed:16431929). Regulates cell growth through regulation of cell cycle progression and cyt
NucleusCytoplasmCytoplasm, cytoskeleton, spindleMidbody
Meckel syndrome 12
A form of Meckel syndrome, a disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Catalyzes the first step in ubiquitin conjugation to mark cellular proteins for degradation through the ubiquitin-proteasome system (PubMed:1447181, PubMed:1606621, PubMed:33108101). Activates ubiquitin by first adenylating its C-terminal glycine residue with ATP, and thereafter linking this residue to the side chain of a cysteine residue in E1, yielding a ubiquitin-E1 thioester and free AMP (PubMed:1447181). Essential for the formation of radiation-induced foci, timely DNA repair and for respon
CytoplasmMitochondrionNucleus
Spinal muscular atrophy X-linked 2
A lethal infantile form of spinal muscular atrophy, a neuromuscular disorder characterized by degeneration of the anterior horn cells of the spinal cord, leading to symmetrical muscle weakness and atrophy. Clinical features include hypotonia, areflexia, and multiple congenital contractures.
Plays a role in interneurons differentiation (PubMed:26056227). Involved in neuronal development and in neuromuscular junction formation
CytoplasmNucleusPostsynaptic cell membrane
Wieacker-Wolf syndrome
A severe X-linked recessive neurodevelopmental disorder affecting the central and peripheral nervous systems. It is characterized by onset of muscle weakness in utero (fetal akinesia). Affected boys are born with severe contractures, known as arthrogryposis, and have delayed motor development, facial and bulbar weakness, characteristic dysmorphic facial features, and skeletal abnormalities, such as hip dislocation, scoliosis, and pes equinovarus. Those that survive infancy show intellectual disability. Carrier females may have mild features of the disorder.
Component of the AP2-containing clathrin coat that may regulate clathrin-dependent trafficking at plasma membrane, TGN and endosomal system (Probable). A possible serine/threonine-protein kinase toward the beta2-subunit of the plasma membrane adapter complex AP2 and other proteins in presence of poly-L-lysine has not been confirmed (PubMed:15809293, PubMed:16914521). By regulating the expression of excitatory receptors at synapses, plays an essential role in neuronal function and signaling and i
Cytoplasmic vesicle, clathrin-coated vesicleGolgi apparatus, trans-Golgi network membraneEndosome membrane
Arthrogryposis multiplex congenita 4, neurogenic, with agenesis of the corpus callosum
A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC4 is an autosomal recessive, severe form with onset in utero. Patients manifest little or no fetal movements, significant contractures affecting the upper and lower limbs, dysmorphic facial features, optic atrophy, limb fractures, profound global developmental delay, seizures, and peripheral neuropathy. Many patients die in early childhood.
Component of the THO subcomplex of the TREX complex which is thought to couple mRNA transcription, processing and nuclear export, and which specifically associates with spliced mRNA and not with unspliced pre-mRNA (PubMed:15833825, PubMed:15998806, PubMed:17190602). Required for efficient export of polyadenylated RNA and spliced mRNA (PubMed:23222130). The THOC1-THOC2-THOC3 core complex alone is sufficient to bind export factor NXF1-NXT1 and promote ATPase activity of DDX39B; in the complex THOC
NucleusNucleus speckleCytoplasm
Intellectual developmental disorder, X-linked, syndromic, Kumar type
A form of intellectual disability, a disorder characterized by significantly below average general intellectual functioning associated with impairments in adaptive behavior and manifested during the developmental period. Intellectual deficiency is the only primary symptom of non-syndromic X-linked forms, while syndromic forms present with associated physical, neurological and/or psychiatric manifestations. MRXSK patients manifest variable degrees of intellectual disability. Commonly observed features included speech delay, elevated BMI, short stature, seizure disorders, gait disturbance, and tremors.
Required for the export of mRNAs containing poly(A) tails from the nucleus into the cytoplasm. May be involved in the terminal step of the mRNA transport through the nuclear pore complex (NPC)
NucleusCytoplasmNucleus, nuclear pore complex
Lethal congenital contracture syndrome 1
A form of lethal congenital contracture syndrome, an autosomal recessive disorder characterized by degeneration of anterior horn neurons, extreme skeletal muscle atrophy, and congenital non-progressive joint contractures (arthrogryposis). The contractures can involve the upper or lower limbs and/or the vertebral column, leading to various degrees of flexion or extension limitations evident at birth. LCCS1 patients manifest early fetal hydrops and akinesia, micrognathia, pulmonary hypoplasia, pterygia, and multiple joint contractures. It leads to prenatal death.
Inhibits fibrillization of amyloid-beta peptide during the elongation phase. Has also been shown to assemble amyloid fibrils into protease-resistant aggregates. Binds heparin
Membrane
Fibrosis of extraocular muscles, congenital, 5
An ocular motility disorder characterized by congenital dysinnervation of various cranial nerves to ocular muscles. Clinical features are ophthalmoplegia, anchoring of the eyes in downward gaze, ptosis, and backward tilt of the head.
Proposed to be involved in endosomal maturation implicating in part VPS33B. In epithelial cells, the VPS33B:VIPAS39 complex may play a role in the apical RAB11A-dependent recycling pathway and in the maintenance of the apical-basolateral polarity (PubMed:20190753). May play a role in lysosomal trafficking, probably via association with the core HOPS complex in a discrete population of endosomes; the functions seems to be independent of VPS33B (PubMed:19109425). May play a role in vesicular traff
CytoplasmCytoplasmic vesicleEarly endosomeRecycling endosomeLate endosome
Arthrogryposis, renal dysfunction and cholestasis syndrome 2
A multisystem disorder, characterized by neurogenic arthrogryposis multiplex congenita, renal tubular dysfunction and neonatal cholestasis with bile duct hypoplasia and low gamma glutamyl transpeptidase activity. Platelet dysfunction is common.
May play a role in vesicle-mediated protein trafficking to lysosomal compartments and in membrane docking/fusion reactions of late endosomes/lysosomes. Required for proper trafficking and targeting of the collagen-modifying enzyme lysyl hydroxylase 3 (LH3) to intracellular collagen (PubMed:28017832). Mediates phagolysosomal fusion in macrophages (PubMed:18474358). Proposed to be involved in endosomal maturation implicating VIPAS39. In epithelial cells, the VPS33B:VIPAS39 complex may play a role
Late endosome membraneLysosome membraneEarly endosomeCytoplasmic vesicle, clathrin-coated vesicleRecycling endosome
Arthrogryposis, renal dysfunction, and cholestasis 1
An autosomal recessive multisystem disorder with characteristics of congenital joint contractures, renal tubular dysfunction, neonatal cholestasis with bile duct hypoplasia and low gamma glutamyl transpeptidase activity, severe failure to thrive, ichthyosis, and a defect in platelet alpha-granule biogenesis. Most patients do not survive past the first year of life.
Component of Schwann cell signaling pathway(s) that controls axon segregation and myelin formation (By similarity)
Secreted
Arthrogryposis multiplex congenita 1, neurogenic, with myelin defect
A form of arthrogryposis multiplex congenita, a developmental condition characterized by multiple joint contractures resulting from reduced or absent fetal movements. AMC1 is an autosomal recessive severe form with onset in utero. Most affected individuals die in utero. Those who survive have generalized contractures and hypotonia. The disorder is caused by a neurogenic defect and poor or absent myelin formation around peripheral nerves rather than by a muscular defect.
PPIases accelerate the folding of proteins during protein synthesis
Endoplasmic reticulum lumen
Osteogenesis imperfecta 11
A form of osteogenesis imperfecta, a disorder of bone formation characterized by low bone mass, bone fragility and susceptibility to fractures after minimal trauma. Disease severity ranges from very mild forms without fractures to intrauterine fractures and perinatal lethality. Extraskeletal manifestations, which affect a variable number of patients, are dentinogenesis imperfecta, hearing loss, and blue sclerae. OI11 is an autosomal recessive form.
Tubulin is the major constituent of microtubules, a cylinder consisting of laterally associated linear protofilaments composed of alpha- and beta-tubulin heterodimers. Microtubules grow by the addition of GTP-tubulin dimers to the microtubule end, where a stabilizing cap forms. Below the cap, tubulin dimers are in GDP-bound state, owing to GTPase activity of alpha-tubulin
Cytoplasm, cytoskeletonCytoplasm, cytoskeleton, flagellum axoneme
Lissencephaly 3
A classic type lissencephaly associated with psychomotor retardation and seizures. Features include agyria or pachygyria or laminar heterotopia, severe intellectual disability, motor delay, variable presence of seizures, and abnormalities of corpus callosum, hippocampus, cerebellar vermis and brainstem.
Plays a role in DNA damage repair as component of the ASCC complex (PubMed:29997253). Part of the ASC-1 complex that enhances NF-kappa-B, SRF and AP1 transactivation (PubMed:12077347). In cells responding to gastrin-activated paracrine signals, it is involved in the induction of SERPINB2 expression by gastrin. May also play a role in the development of neuromuscular junction
NucleusNucleus speckle
Barrett esophagus
A condition characterized by a metaplastic change in which normal esophageal squamous epithelium is replaced by a columnar and intestinal-type epithelium. Patients with Barrett esophagus have an increased risk of esophageal adenocarcinoma. The main cause of Barrett esophagus is gastroesophageal reflux. The retrograde movement of acid and bile salts from the stomach into the esophagus causes prolonged injury to the esophageal epithelium and induces chronic esophagitis, which in turn is believed to trigger the pathologic changes.
Probable adhesion protein, which mediates homophilic and heterophilic interactions. In contrast to SCARF1, it poorly mediates the binding and degradation of acetylated low density lipoprotein (Ac-LDL) (By similarity)
Membrane
Van den Ende-Gupta syndrome
A syndrome characterized by craniofacial and skeletal abnormalities that include blepharophimosis, a flat and wide nasal bridge, narrow and beaked nose, hypoplastic maxilla with or without cleft palate and everted lower lip, prominent ears, down-slanting eyes, arachnodactyly, and camptodactyly. Patients present congenital joint contractures that improve without intervention, and normal growth and development. Intelligence is normal. Rarely, enlarged cerebella can be present. Some patients experience respiratory problems due to laryngeal abnormalities.
Pore-forming subunit of the mechanosensitive non-specific cation Piezo channel required for rapidly adapting mechanically activated (MA) currents and has a key role in sensing touch and tactile pain (PubMed:37590348). Piezo channels are homotrimeric three-blade propeller-shaped structures that utilize a cap-motion and plug-and-latch mechanism to gate their ion-conducting pathways (PubMed:37590348). Expressed in sensory neurons, is essential for diverse physiological processes, including respirat
Cell membrane
Arthrogryposis, distal, 5
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA5 features include ocular abnormalities, typically ptosis, ophthalmoplegia and/or strabismus, in addition to contractures of the skeletal muscles. Some patients have pulmonary hypertension as a result of restrictive lung disease.
Component of nuclear pore complex
Nucleus, nuclear pore complex
Fetal akinesia deformation sequence 4
A clinically and genetically heterogeneous group of disorders with congenital malformations related to impaired fetal movement. Clinical features include fetal akinesia, intrauterine growth retardation, polyhydramnios, arthrogryposis, pulmonary hypoplasia, craniofacial abnormalities, and cryptorchidism. FADS4 inheritance is autosomal recessive.
Probably enhances ubiquitin ligase activity of RING-type zinc finger-containing E3 ubiquitin-protein ligases, possibly through recruitment and/or stabilization of the Ubl-conjugating enzyme (E2) at the E3:substrate complex. Acts as a regulator of retrograde transport via its interaction with VPS35. Recruited to retromer-containing endosomes and promotes the formation of 'Lys-63'-linked polyubiquitin chains at 'Lys-220' of WASHC1 together with TRIM27, leading to promote endosomal F-actin assembly
Early endosomeCytoplasmNucleus
Schaaf-Yang syndrome
A disease characterized by clinical features of Prader-Willi syndrome, including neonatal hypotonia with poor suck, feeding problems in infancy, obesity, developmental delay, short stature, and hypogonadism. Additionally, patients manifest autism spectrum disorder. Some patients have dysmorphic facial features.
Possible role in transport between endoplasmic reticulum and Golgi
Endoplasmic reticulum membraneEndoplasmic reticulum-Golgi intermediate compartment membraneGolgi apparatus membrane
Arthrogryposis multiplex congenita 2, neurogenic type
A form of arthrogryposis multiplex congenita, a heterogeneous group of disorders characterized by multiple joint contractures resulting, in some cases, from reduced or absent fetal movements. AMC2 is due to a neurogenic defect and is characterized by congenital immobility of the limbs with fixation of multiple joints, and muscle wasting. AMC2 transmission pattern is consistent with autosomal recessive inheritance in several families. Penetrance may be incomplete in females.
Receptor tyrosine kinase which plays a central role in the formation and the maintenance of the neuromuscular junction (NMJ), the synapse between the motor neuron and the skeletal muscle (PubMed:25537362). Recruitment of AGRIN by LRP4 to the MUSK signaling complex induces phosphorylation and activation of MUSK, the kinase of the complex. The activation of MUSK in myotubes regulates the formation of NMJs through the regulation of different processes including the specific expression of genes in s
Postsynaptic cell membrane
Myasthenic syndrome, congenital, 9, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS9 is a disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Electrogenic antiporter that exchanges one cholinergic neurotransmitter, acetylcholine or choline, with two intravesicular protons across the membrane of synaptic vesicles. Uses the electrochemical proton gradient established by the V-type proton-pump ATPase to store neurotransmitters inside the vesicles prior to their release via exocytosis (By similarity) (PubMed:20225888, PubMed:8910293). Determines cholinergic vesicular quantal size at presynaptic nerve terminals in developing neuro-muscular
Cytoplasmic vesicle, secretory vesicle, synaptic vesicle membrane
Myasthenic syndrome, congenital, 21, presynaptic
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness. CMS21 is an autosomal recessive, pre-synaptic form characterized by ptosis, ophthalmoplegia, fatigable weakness, apneic crises, and deterioration of symptoms in cold water. Learning difficulties and left ventricular dysfunction may be present in some patients.
Transcription coactivator which associates with nuclear receptors, transcriptional coactivators including EP300, CREBBP and NCOA1, and basal transcription factors like TBP and TFIIA to facilitate nuclear receptors-mediated transcription (PubMed:10454579, PubMed:25219498). May thereby play an important role in establishing distinct coactivator complexes under different cellular conditions (PubMed:10454579, PubMed:25219498). Plays a role in thyroid hormone receptor and estrogen receptor transactiv
NucleusCytoplasm, cytosolCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Spinal muscular atrophy with congenital bone fractures 1
An autosomal recessive neuromuscular disorder characterized by prenatal-onset spinal muscular atrophy, multiple congenital contractures consistent with arthrogryposis multiplex congenita, respiratory distress, and congenital bone fractures.
Acts as a transcriptional activator that promotes transcription of muscle-specific target genes and plays a role in muscle differentiation. Together with MYF5 and MYOG, co-occupies muscle-specific gene promoter core region during myogenesis. Induces fibroblasts to differentiate into myoblasts. Interacts with and is inhibited by the twist protein. This interaction probably involves the basic domains of both proteins (By similarity)
Nucleus
Congenital myopathy 17
An autosomal recessive muscular disorder characterized by hypotonia and respiratory insufficiency apparent soon after birth, high diaphragmatic dome on imaging, poor overall growth, pectus excavatum, dysmorphic facies, and renal anomalies in some affected individuals. Additional variable features include delayed motor development, mildly decreased endurance, distal arthrogryposis, and lung hypoplasia resulting in early death.
This giant muscle protein may be involved in maintaining the structural integrity of sarcomeres and the membrane system associated with the myofibrils. Binds and stabilize F-actin
Cytoplasm, myofibril, sarcomereCytoplasm, cytoskeleton
Nemaline myopathy 2
A form of nemaline myopathy. Nemaline myopathies are muscular disorders characterized by muscle weakness of varying severity and onset, and abnormal thread-like or rod-shaped structures in muscle fibers on histologic examination.
Postsynaptic protein required for clustering of nicotinic acetylcholine receptors (nAChRs) at the neuromuscular junction. It may link the receptor to the underlying postsynaptic cytoskeleton, possibly by direct association with actin or spectrin
Cell membranePostsynaptic cell membraneCytoplasm, cytoskeleton
Myasthenic syndrome, congenital, 11, associated with acetylcholine receptor deficiency
A form of congenital myasthenic syndrome, a group of disorders characterized by failure of neuromuscular transmission, including pre-synaptic, synaptic, and post-synaptic disorders that are not of autoimmune origin. Clinical features are easy fatigability and muscle weakness affecting the axial and limb muscles (with hypotonia in early-onset forms), the ocular muscles (leading to ptosis and ophthalmoplegia), and the facial and bulbar musculature (affecting sucking and swallowing, and leading to dysphonia). The symptoms fluctuate and worsen with physical effort. CMS11 is an autosomal recessive disorder of postsynaptic neuromuscular transmission, due to deficiency of AChR at the endplate that results in low amplitude of the miniature endplate potential and current.
Variantes genéticas (ClinVar)
159 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.086 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
84 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Artrogripose múltipla congênita
Centros de Referência SUS
24 centros habilitados pelo SUS para Artrogripose múltipla congênita
Centros para Artrogripose múltipla congênita
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
19 ensaios clínicos encontrados, 5 ativos.
Publicações mais relevantes
Management of Lower-Extremity Deformity in Arthrogryposis Multiplex Congentia: A Narrative Review.
Arthrogryposis multiplex congenita (AMC) is a highly heterogeneous constellation of disorders defined by non-progressive congenital multiple joint contractures, typically manifesting across multiple limbs. The pathology results in significant functional impairment, including restricted range of motion, chronic arthralgia, and secondary musculoskeletal deformities like scoliosis. Given that AMC is an umbrella designation encompassing over 300 distinct etiologies, the profound clinical variability poses substantial diagnostic and therapeutic challenges. The relative rarity of AMC and the absence of consensus-based, longitudinal treatment protocols create a critical void in standardized clinical management across the lifespan of patients. This comprehensive narrative review synthesizes the contemporary literature and clinical evidence to establish a structured, life-course management paradigm, extending from neonatal screening through to adult care. We advocate for an evidence-based approach that recalibrates therapeutic goals to emphasize maximal functional capacity and societal participation rather than strict anatomical normalization. Key aspects addressed include early-life neuroplasticity, the principles of staged and minimally- invasive surgical correction, and the need for seamless, lifelong, multidisciplinary care coordination. Furthermore, the review critically examines persistent clinical dilemmas concerning hip and knee contracture management, and proposes algorithmic pathways for addressing recurrent foot deformities. By integrating the latest advancements in molecular genetics, surgical innovations, and rehabilitative science, this work serves as an authoritative resource, offering clinically applicable strategies to optimize long-term outcomes for individuals living with AMC.
Arthrogryposis as a neuromuscular phenotype: lessons from PIEZO2 loss-of-function.
Arthrogryposis multiplex congenita is a heterogeneous group of disorders characterised by multiple joint contractures resulting from impaired fetal movement. A 43-year-old woman presented with a long-standing congenital clubfoot, progressive scoliosis since childhood and distal joint contractures. She had pure sensory findings, with global areflexia, severe sensory ataxia and a positive Romberg sign, significantly affecting her balance and gait. Genetic analysis identified two likely pathogenic variants in the PIEZO2 gene, consistent with an autosomal recessive inheritance pattern. We discuss the differential diagnosis of arthrogryposis multiplex congenita, emphasising the importance of considering neuromuscular causes. This case highlights the role of comprehensive neurological assessment in patients with distal joint contractures and scoliosis, showing that detailed peripheral neurological findings can guide genetic testing and lead to an accurate diagnosis.
CRISPR Activation Reveals the Spliceogenicity of an Intronic NEB Variant in Fetuses With Arthrogryposis Multiplex Congenita 6.
Whole-genome sequencing identifies intronic variants whose pathogenicity can be predicted with tools like SpliceAI. However, an actionable classification of such variants may require RNA-based validation, which can be limited by low expression in clinically accessible tissues. We report two fetuses from one family with Arthrogryposis multiplex congenita 6 (AMC6 [OMIM # 619334]) and biallelic NEB variants: a paternally inherited likely pathogenic frameshift variant, Chr2(GRCh38):g.151579391del; NM_001164508.2:c.16653del; NP_001157980.2:p.(Asp5552ThrfsTer5), and a maternally inherited intronic variant of uncertain clinical significance, Chr2(GRCh38):g.151496267G>A; NM_001164508.2:c.24486+9C>T; NP_001157980.2:p.(?). Because NEB is poorly expressed in fibroblasts, we used CRISPR activation to induce its expression in fibroblasts from the heterozygous mother. RNA-sequencing subsequently confirmed that the intronic variant generated a novel splice donor site associated with inferred loss of splicing at the canonical donor site. After NMD-inhibition, we could thus identify 45.5% of NEB transcripts with a 7 bp exon extension, predicted to result in a protein-coding frameshift. The intronic variant was classified as likely pathogenic, allowing a genetic diagnosis.
Perinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
Arthrogryposis multiplex congenita (AMC) presents challenges for prenatal detection due to its heterogeneous etiology, onset, and phenotypical manifestations. This study aims to describe the genetic diagnostic yield in a population of fetuses with detailed phenotypic description over a 15-year period (2007-2021) at the Fetal Medicine Unit of Amsterdam UMC, the Netherlands. The fetal and neonatal phenotypes were classified into three clinical AMC Groups, with the exception that Groups 1 and 2 were combined in the prenatal classification. Group 1 involves limb involvement primarily, Group 2 includes musculoskeletal involvement plus other system anomalies, and Group 3 involves musculoskeletal involvement with central nervous system disability, lethality, fetal akinesia deformation sequence, and/or intellectual disability. The cohort consisted of 64 consecutive cases, 13 in Groups 1 + 2 and 51 in Group 3. Perinatal genetic testing occurred in all cases: prenatally in 56 of the 64 (88%), postnatally in 36 of the 64 (56%), and combined testing in 28 of the 64 cases (44%). The overall genetic diagnostic yield was 28% (18/64), and it increased over the 5-year period from 14% to 50%. Whole exome sequencing had the highest yield (41.7%). The yield per phenotype was 30.8% (4/13) for AMC Group 1 + 2 and 27.4% (14/51) for AMC Group 3. Detailed fetal phenotyping and perinatal genetic testing in all cases showed improved diagnostic yield over time, likely due to the introduction of Next-generation sequencing-based tests. The availability of stored DNA will be beneficial for future investigations since further improvements in genetic testing possibilities are expected.
Expanding the Phenotypic Spectrum of Syndromic Arthrogryposis Multiplex Congenita: Role of Biallelic Variants in COL25A1 Across Fetal and Pediatric Periods.
Publicações recentes
Prenatal Diagnosis of Arthrogryposis Multiplex Congenita (AMC): Ultrasound and Genetic Findings in 69 Fetuses From 67 Unrelated Families.
Growth arrest following posterior knee release in children with arthrogryposis multiplex congenita: a previously unreported complication.
Milestone Attainment in Young Children With Arthrogryposis Multiplex Congenita: Developmental Profile and Associated Factors.
Management of Lower-Extremity Deformity in Arthrogryposis Multiplex Congentia: A Narrative Review.
Expanding the Phenotypic Spectrum of Syndromic Arthrogryposis Multiplex Congenita: Role of Biallelic Variants in COL25A1 Across Fetal and Pediatric Periods.
📚 EuropePMC542 artigos no totalmostrando 199
Growth arrest following posterior knee release in children with arthrogryposis multiplex congenita: a previously unreported complication.
Journal of pediatric orthopedics. Part BMilestone Attainment in Young Children With Arthrogryposis Multiplex Congenita: Developmental Profile and Associated Factors.
American journal of medical genetics. Part C, Seminars in medical geneticsManagement of Lower-Extremity Deformity in Arthrogryposis Multiplex Congentia: A Narrative Review.
Medical science monitor : international medical journal of experimental and clinical researchExpanding the Phenotypic Spectrum of Syndromic Arthrogryposis Multiplex Congenita: Role of Biallelic Variants in COL25A1 Across Fetal and Pediatric Periods.
Indian journal of pediatricsGenetic Bases of Arthrogryposis Multiplex Congenita.
Annual review of genomics and human geneticsSurgical timing and management patterns across joints in arthrogryposis: a systematic review.
Journal of pediatric orthopedics. Part BPreemptive immunotherapy for fetal acetylcholine receptor antibody disorder (FARAD): from recurrent pregnancy losses to a healthy infant - a case report.
Neuromuscular disorders : NMDTibialis Anterior Tendon Transfer in an Arthrogrypotic Clubfoot: A Case Report.
JBJS case connectorPediatric patients with arthrogryposis have increased early complications and long-term reoperation risk following posterior spinal fusion.
Spine deformityArthrogryposis as a neuromuscular phenotype: lessons from PIEZO2 loss-of-function.
Practical neurologyFactors Associated With Functional Mobility in 256 Children With Arthrogryposis Multiplex Congenita: A Multicentric Cross-Sectional Study.
Archives of physical medicine and rehabilitationP18 ZMPSTE24 variant with the lethal phenotype of restrictive dermopathy.
The British journal of dermatologyGross motor functional classification for arthrogryposis multiplex congenita: protocol for co-development involving public with lived and professional experience.
Research involvement and engagementPierre Robin Sequence Associated with Arthrogryposis Multiplex Congenita.
Indian journal of pediatricsPsychometric properties of functional mobility outcome measures in children with arthrogryposis multiplex congenita.
Developmental medicine and child neurologyHydrops, Arthrogryposis, and Cerebellar Hypoplasia in a Fetus With a de Novo BICD2 Variant: Expanding the Prenatal Phenotype of SMALED2B.
Prenatal diagnosisBiallelic variants in the UTRN gene cause a novel form of multiple congenital arthrogryposis.
Frontiers in geneticsVariant Update on ASCC1 : Characterization of the First Homozygous Missense Variant Involved in Prenatal-Onset Spinal Muscular Atrophy With Congenital Bone Fractures 2.
American journal of medical genetics. Part AFracture Risk Following Hardware Removal in Children With Arthrogryposis.
Journal of pediatric orthopedicsCRISPR Activation Reveals the Spliceogenicity of an Intronic NEB Variant in Fetuses With Arthrogryposis Multiplex Congenita 6.
Clinical geneticsSENP7-Related Fatal Arthrogryposis Multiplex Congenita and Immunodeficiency: A Novel Missense Variation and Syndromic Delineation.
Clinical geneticsAnesthetic management of children undergoing specialized orthopedic surgeries.
Journal of clinical orthopaedics and traumaBiallelic CRELD1 variants cause severe muscle weakness and infantile epilepsy.
Brain communications[Expert consensus on the clinical diagnosis and management of Arthrogryposis multiplex congenita (2025 Edition)].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsThumb-in-palm deformity in arthrogryposis multiplex congenita: A severity-based classification and structured surgical approach.
Journal of clinical orthopaedics and traumaContent validity of mobility measures in arthrogryposis multiplex congenita: engaging clinicians and people with lived experience.
Frontiers in rehabilitation sciencesLong-term Function of Adults With Arthrogryposis.
Journal of pediatric orthopedicsADGRG6-related disorder: a novel mutation resulting in distal arthrogryposis and a patchy neuropathy.
Neuromuscular disorders : NMDArthrogryposis Multiplex Congenita (AMC) and counselling before and during pregnancy: a questionnaire study.
Orphanet journal of rare diseasesMaternal experience of fetal movements from a child with AMC: MECA survey.
Early human developmentFetal Akinesia/Hypokinesia and Arthrogryposis of Neuromuscular Origin: Etiologic Groups, Genetics, and Phenotypic Spectrum.
Annals of clinical and translational neurologyArthrogryposis Multiplex Congenita Discovered at Birth: A Case Report.
CureusPathogenic variants in BORCS5 Cause a Spectrum of Neurodevelopmental and Neurodegenerative Disorders with Lysosomal Dysfunction.
medRxiv : the preprint server for health sciencesThe evolving genetic landscape of neuromuscular fetal akinesias.
Journal of neuromuscular diseasesDoes Open Reduction of Arthrogrypotic Hips Cause Stiffness?
Journal of pediatric orthopedicsFrom abnormal fetal movements to neonatal seizures: A literature review.
Epilepsy researchNovel SCYL2 Mutations and Arthrogryposis Multiplex Congenita 4: Case Report and Review of the Literature.
International journal of molecular sciencesBiallelic Variant, c.644-13_644-9del in UNC50 Is Associated With Congenital Myasthenia Syndrome.
American journal of medical genetics. Part AConsensus-based recommendations for the rehabilitation of children with arthrogryposis multiplex congenita: an integrated knowledge translation approach.
Orphanet journal of rare diseasesOpen Reduction of Hip Dislocation Is Associated with Higher Rates of Proximal Femoral Growth Disturbance in Patients with Arthrogryposis Multiplex Congenita Than Idiopathic DDH: A Dual-Center Retrospective Cohort Study.
The Journal of bone and joint surgery. American volumeAnesthetic Management of Inguinal Hernia Surgery in an Adult Patient With Arthrogryposis Multiplex Congenita: A Case Report.
CureusPerinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
European journal of human genetics : EJHGThe First Known Case Report of a Novel Homozygous Nonsense Variant in the OSBPL9 Gene Associated With Fetal Cerebral Ventriculomegaly, Cerebellar Hypoplasia, and Arthrogryposis Multiplex.
CureusHuman Phenotype Ontology Annotations for Rare Congenital Conditions: Application to Arthrogryposis Multiplex Congenita.
American journal of medical genetics. Part APrenatal Diagnosis and Prognostic Factors in Fetuses With Arthrogryposis Multiplex Congenita-A Systematic Review.
Journal of clinical ultrasound : JCUEvaluation of Foot Osteotomies for Treating Residual Clubfoot Deformities in Ambulatory Children With Arthrogryposis.
Journal of pediatric orthopedicsMaternal, fetal and neonatal outcomes among pregnant women with arthrogryposis multiplex congenita: a scoping review.
Orphanet journal of rare diseasesStaged Correction of Hip Contractures, Severe Knee Flexion, and Clubfoot in Arthrogryposis: Enabling Assisted Ambulation.
JBJS case connectorVerbal fluency and semantic association deficits in children with in birth nonprogressive neuromuscular diseases.
Frontiers in human neuroscienceNeurogenic arthrogryposis, hypotonia, dysmorphic features plus malformation of cortical development further expands the ARL6IP1 loss-of-function phenotype.
Neuromuscular disorders : NMDMaintained gait in persons with arthrogryposis from childhood to adulthood.
BMC musculoskeletal disordersIdentification of novel RIPK4 variants in a Chinese patient with Arthrogryposis Multiplex Congenita (AMC).
Italian journal of pediatricsTranscriptional Changes Associated with Amyoplasia.
International journal of molecular sciencesSensory and Motor Function, Pain, and Health Status in Children with Arthrogryposis and Myelomeningocele.
Children (Basel, Switzerland)Lethal Phenotype and Expansion of the Clinical Spectrum of Biallelic Loss of Function Variant in SENP7 Gene Unveiled by Whole Exome Sequencing.
Clinical geneticsA Splice Site Variant in SENP7 Results in a Severe Form of Arthrogryposis.
Clinical geneticsECEL1 mutation in distal arthrogryposis type 5D: A case report.
European journal of obstetrics, gynecology, and reproductive biologyThe First Case Report of a Homozygous Consensus Acceptor Splice Variant in the NUP214 Gene Associated With Fetal Hydrops and Arthrogryposis Multiplex.
Cureus[Arthrogryposis Multiplex Congenita in pediatric age: Correlation between Muscle MRI and functional assessment].
Medecine sciences : M/SHealth-related quality of life in 205 children with arthrogryposis multiplex congenita.
Quality of life research : an international journal of quality of life aspects of treatment, care and rehabilitationDo Not Forget the Spine MRI in Children With Arthrogryposis Multiplex Congenita: High Prevalence of Tethered Spinal Cord and Preliminary Clinical Findings Following Detethering.
Journal of pediatric orthopedicsLoss of ZC4H2, an Arthrogryposis Multiplex Congenita Associated Gene, Promotes Osteoclastogenesis in Mice.
GenesSCYL2-related autosomal recessive neurodevelopmental disorders: Arthrogryposis multiplex congenita-4 and beyond?
Clinical geneticsGenotype-Phenotype Correlations and Sex Differences in ZC4H2-Associated Rare Disorder.
Pediatric neurologyCongenital Contractures and Fractures: A Variant of Bruck Syndrome Type 2.
CureusExpansion of the phenotypic spectrum of KARS1-related disorders to include arthrogryposis multiplex congenita and summary of experiences with lysine supplementation.
American journal of medical genetics. Part AEarly Diagnosis and Management of Arthrogryposis Multiplex Congenita in a Neonate: A Case Study.
CureusThe experience of caregiving for children with rare musculoskeletal conditions: a qualitative study in arthrogryposis multiplex congenita.
Orphanet journal of rare diseasesMultiple Pterygium Syndrome (Escobar Syndrome): A Rare Form of Prenatal Myasthenia Presenting With Arthrogryposis Multiplex Congenita.
NeurologySTAC3 disorder: a common cause of congenital hypotonia in Southern African patients.
European journal of human genetics : EJHGAn Unusual Presentation of Membranous Nephropathy in an Adult With Arthrogryposis: A Case Report.
CureusThe Intrarater and Interrater Reliability of the OMT Classification Among Physicians With a Different Background.
Journal of pediatric orthopedicsArthrogryposis multiplex congenita causing hypercapnic respiratory failure and airway obstruction in an adult patient.
Respirology case reportsBone mineral density in adults with arthrogryposis multiplex congenita: a retrospective cohort analysis.
Scientific reportsPrenatal diagnosis (or lack thereof) of arthrogryposis multiplex congenita and its impact on the perinatal experience of parents: A retrospective survey.
Prenatal diagnosisBilateral Sprengel Deformities, Mirror Movements Synkinesis, and Arthrogryposis Multiplex Congenita: A Novel Combination.
Journal of orthopaedic case reportsSubcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf-Yang syndrome.
Journal of medical genetics[Nerve Transfers in Children with Non-traumatic Amyoplasia].
Handchirurgie, Mikrochirurgie, plastische Chirurgie : Organ der Deutschsprachigen Arbeitsgemeinschaft fur Handchirurgie : Organ der Deutschsprachigen Arbeitsgemeinschaft fur Mikrochirurgie der Peripheren Nerven und Gefasse : Organ der V...Multiple Serial Casting for Recurrent Clubfoot in Arthrogryposis Corrects Deformity With Diminishing Returns.
Cureus[Myasthenia gravis-Gender aspects and family planning].
Der NervenarztPregnancy in myasthenia gravis: a retrospective analysis of maternal and neonatal outcome from a large tertiary care centre in Germany.
Archives of gynecology and obstetricsCommon data elements for arthrogryposis multiplex congenita: An international framework.
Developmental medicine and child neurologyArthrogryposis Multiplex Congenita and the Importance of Orthoses: A Case Report.
CureusA Case Report: Can Prioritizing Sensory Integration Therapy Help Improve Gross Motor Function in a Rare Case of Neurogenic Arthrogryposis Multiplex Congenita?
CureusCompound heterozygous variants in MYBPC1 lead to severe distal arthrogryposis type-1 manifestations.
GeneThe science of uncertainty guides fetal-neonatal neurology principles and practice: diagnostic-prognostic opportunities and challenges.
Frontiers in neurologySimple Talectomy is a Beneficial Surgical Procedure for Talipes Equinovarus and Other Severe Neuromuscular Foot Deformities.
The Journal of foot and ankle surgery : official publication of the American College of Foot and Ankle SurgeonsBiallelic truncating TTN variants in M-band encoding exons cause a fetal lethal titinopathy.
Prenatal diagnosisArthrogryposis multiplex congenita: dental and maxillofacial phenotype - A scoping review.
BoneMuscular phenotype description of abnormal THOC2 splicing.
Neuromuscular disorders : NMDPerspectives from clinicians and managers: facilitators and barriers to the uptake of rehabilitation guidance for children with arthrogryposis.
Disability and rehabilitationA rare case of arthrogryposis multiplex congenita in a 2-year-old boy case report.
SAGE open medical case reportsStakeholder engagement in the development of an upper extremity outcome measure for children with rare musculoskeletal conditions.
Research involvement and engagementSpinal Fusion in Patients With Classic Amyoplasia and General Arthrogryposis.
Journal of pediatric orthopedicsNovel TTN Mutation Causing Severe Congenital Myopathy and Uncertain Association with Infantile Hydrocephalus.
Case reports in geneticsUnusual Presentation of Pediatric Scurvy: A Necrotic Gastrostomy Tube Site in a 14-Year-Old Boy.
The American journal of case reportsExome sequencing links the SUMO protease SENP7 with fatal arthrogryposis multiplex congenita, early respiratory failure and neutropenia.
Journal of medical geneticsNeonatal developmental and epileptic encephalopathy with movement disorders and arthrogryposis: A case report with a novel missense variant of SCN1A.
Brain & developmentDST variants are responsible for neurogenic arthrogryposis multiplex congenita enlarging the spectrum of type VI hereditary sensory autonomic neuropathy.
Clinical geneticsMutation In Fkbp10 Gene Cause Bruck Syndrome 1 (Brks1) In A Pakistani Family Of Pashtun Origin.
Journal of Ayub Medical College, Abbottabad : JAMCGrowth Charts for Children With Arthrogryposis Multiplex Congenita.
Clinical pediatrics[Comparison of intraoperative neurophysiological monitoring between patients with arthrogryposis multiplex congenita and adolescent idiopathic scoliosis].
Zhonghua yi xue za zhiSurvival in Mudejar Spain in the Middle Ages (thirteenth-fourteenth centuries): Ancient Rare Diseases-an uncommon diagnosis in archaeological human remains.
International orthopaedicsMeasurement Repeatability and Reproducibility of Virtual Goniometry of a Set of Acquired Images in Youths with Arthrogryposis Multiplex Congenita.
Journal of musculoskeletal & neuronal interactionsRehabilitation in Patients Diagnosed with Arthrogryposis Multiplex Congenita: A Systematic Review.
Children (Basel, Switzerland)Effectiveness of Ponseti technique in management of arthrogrypotic clubfeet - a prospective study.
International journal of burns and traumaThe emerging spectrum of fetal acetylcholine receptor antibody-related disorders (FARAD).
Brain : a journal of neurologyMental health in adults living with arthrogryposis multiplex congenita.
American journal of medical genetics. Part C, Seminars in medical geneticsThe range of publications on arthrogryposis multiplex congenita from 1995 to 2022-A scoping review.
American journal of medical genetics. Part AHomozygous loss-of-function variants in FILIP1 cause autosomal recessive arthrogryposis multiplex congenita with microcephaly.
Human geneticsEye-tracking visual patterns of sonographers with and without fetal motor assessment expertise.
Early human developmentSimilar Cognitive Skill Impairment in Children with Upper Limb Motor Disorders Due to Arthrogryposis Multiplex Congenita and Obstetrical Brachial Plexus Palsy.
International journal of environmental research and public healthThe clinical and genetic spectrum of autosomal-recessive TOR1A-related disorders.
Brain : a journal of neurologyArthrogryposis multiplex congenita with maxillofacial involvement: a case report.
Maxillofacial plastic and reconstructive surgeryNovel Arthrogryposis Multiplex Congenita Presentation in a Newborn With Pierpont Syndrome.
Journal of investigative medicine high impact case reportsSkeletal anomaly and opisthotonus in early-onset epileptic encephalopathy with KCNQ2 abnormality.
Brain & developmentThe Effectiveness of Serial Casting in the Treatment of Recurrent Equinovarus in Children With Arthrogryposis.
Journal of pediatric orthopedicsFetal arthrogryposis-what do we tell the prospective parents?
Prenatal diagnosisCurrent rehabilitation practice for the evaluation and treatment of children with arthrogryposis: an international survey.
Disability and rehabilitationAltered Cerebral Processing of Videos in Children with Motor Dysfunction Suggests Broad Embodiment of Perceptual Cognitive Functions.
Journal of personalized medicineFetal akinesia deformation sequence syndrome associated with recessive TTN variants.
American journal of medical genetics. Part AEarly onset hereditary neuronopathies: an update on non-5q motor neuron diseases.
Brain : a journal of neurologyAltered evoked responses for motor-related words in children with upper limb motor impairments.
Clinical neurophysiology : official journal of the International Federation of Clinical NeurophysiologyRapid genome sequencing identifies a novel de novo SNAP25 variant for neonatal congenital myasthenic syndrome.
Cold Spring Harbor molecular case studiesEpidemiology, aetiology, interventions and genomics in children with arthrogryposis multiplex congenita: protocol for a multisite registry.
BMJ openTalectomy for arthrogrypotic foot deformities: A systematic review.
Foot and ankle surgery : official journal of the European Society of Foot and Ankle SurgeonsArthrogryposis multiplex congenita in a child with congenital fractures: a case report.
Journal of medical case reportsLoss of Protein Function Causing Severe Phenotypes of Female-Restricted Wieacker Wolff Syndrome due to a Novel Nonsense Mutation in the ZC4H2 Gene.
GenesA reverse genetics and genomics approach to gene paralog function and disease: Myokymia and the juxtaparanode.
American journal of human geneticsCase Report: Prenatal Diagnosis of Nemaline Myopathy.
Frontiers in pediatricsDistal Arthrogryposis type 5 in an Italian family due to an autosomal dominant gain-of-function mutation of the PIEZO2 gene.
Italian journal of pediatricsLethal Congenital Contracture Syndrome 11: A Case Report and Literature Review.
Journal of clinical medicineCommentary on "Characterizing Pain Among Adolescents and Young Adults With Arthrogryposis Multiplex Congenita".
Pediatric physical therapy : the official publication of the Section on Pediatrics of the American Physical Therapy AssociationProgressive Respiratory Insufficiency in a Teenager with Diaphragmatic Hypomotility Due to a Novel Combination of Gliomedin Gene Variants.
Children (Basel, Switzerland)First report of SYNE1 arthrogryposis multiplex congenita from Saudi Arabia with a novel mutation: a case report.
Italian journal of pediatricsDisability and Quality of Life After Talectomy for Arthrogryposis Multiplex Congenita.
Foot & ankle internationalBroadening the phenotypic spectrum of TUBA1A tubulinopathy to syndromic arthrogryposis multiplex congenita.
American journal of medical genetics. Part AAbductor pollicis brevis rerouting and first web deepening for clasped thumb deformity in arthrogryposis multiplex congenita.
The Journal of hand surgery, European volumeCharacterizing Pain Among Adolescents and Young Adults With Arthrogryposis Multiplex Congenita.
Pediatric physical therapy : the official publication of the Section on Pediatrics of the American Physical Therapy AssociationRecognizable Pattern of Arthrogryposis and Congenital Myopathy Caused by the Recurrent TTN Metatranscript-only c.39974-11T > G Splice Variant.
NeuropediatricsLong-term follow-up of a patient with autosomal dominant lower extremity-predominant spinal muscular atrophy-2 due to a BICD2 variant.
Brain & developmentA novel ZC4H2 variant in a female with severe respiratory complications.
Brain & developmentA very rare cause of arthrogryposis multiplex congenita: a novel mutation in TOR1A.
Journal of pediatric endocrinology & metabolism : JPEMCase Report: Late-Onset Autosomal Recessive Cerebellar Ataxia Associated With SYNE1 Mutation in a Chinese Family.
Frontiers in geneticsCongenital Dislocation of the Knee: Idiopathic or Arthrogryposis?
CureusExpert guidance for the rehabilitation of children with arthrogryposis: protocol using an integrated knowledge translation approach.
Research involvement and engagementArt and Paediatric Orthopaedics: A Sixteenth Century Lie-down Comedian.
Journal of pediatric orthopedicsTest-retest reliability for performance-based outcome measures among individuals with arthrogryposis multiplex congenita.
BMC musculoskeletal disordersRecessive variants in COL25A1 gene as novel cause of arthrogryposis multiplex congenita with ocular congenital cranial dysinnervation disorder.
Human mutationClinical and Genetic Findings in a Series of Eight Families with Arthrogryposis.
GenesLong-term follow-up of children with a surgically treated clubfoot: Assessing the multi-segment-foot motions, dynamic plantar pressures, and functional outcomes.
Journal of clinical orthopaedics and traumaAnaesthetic management in a patient with arthrogryposis multiplex congenita.
Revista espanola de anestesiologia y reanimacionA woman in her fifties with chronic muscle weakness.
Tidsskrift for den Norske laegeforening : tidsskrift for praktisk medicin, ny raekkeNovel Consensus Splice Site Pathogenic Variation in THOC2 Gene Leads to Recurrent Arthrogryposis Multiplex Congenita Phenotype: A Case Report.
CureusDiagnostic workup in children with arthrogryposis: description of practices from a single reference centre, comparison with literature and suggestion of recommendations.
Journal of medical geneticsLocomotion and Topographical Working Memory in Children With Myelomeningocele and Arthrogryposis Multiplex Congenita.
Frontiers in psychiatryPsychosocial wellbeing among children and adults with arthrogryposis: a scoping review.
Health and quality of life outcomesComparative evaluation and analysis of outcomes in non-idiopathic and idiopathic clubfeet with Ponseti method at a tertiary care centre of a developing country.
Foot (Edinburgh, Scotland)Care Pathway for Foetal Joint Contractures, Foetal Akinesia Deformation Sequence, and Arthrogryposis Multiplex Congenita.
Fetal diagnosis and therapyFalls and Associated Factors among Adolescents and Young Adults with Arthrogryposis Multiplex Congenita.
International journal of rare diseases & disordersImprovement of Pulmonary Function in Arthrogryposis Multiplex Congenita Patients Undergoing Posterior Spinal Fusion Surgery for Concomitant Scoliosis: A Minimum of 3-Year Follow-up.
World neurosurgeryFeasibility and Challenges of Performing Magnetoencephalography Experiments in Children With Arthrogryposis Multiplex Congenita.
Frontiers in pediatricsArthrogryposis multiplex congenita: a 28-year retrospective study.
Developmental medicine and child neurologyWieacker-Wolff syndrome, a distinctive phenotype of arthrogryposis multiplex congenita caused by a "de novo" ZC4H2 gene partial deletion.
Clinical case reportsUltrasonographic measurement of the dimensions of proximal and distal patellar fragments after Niebauer-King Procedure for the management of congenital dislocation of the knee.
Acta orthopaedica et traumatologica turcicaThe Clinical and Genotypic Spectrum of Scoliosis in Multiple Pterygium Syndrome: A Case Series on 12 Children.
GenesPostnatal Diagnostic Workup in Children With Arthrogryposis: A Series of 82 Patients.
Journal of child neurologyPatient-Reported Outcome Measurement Information System (PROMIS) Scores in Pediatric Patients With Arthrogryposis.
Journal of pediatric orthopedicsA Genomic Approach to Delineating the Occurrence of Scoliosis in Arthrogryposis Multiplex Congenita.
GenesNeuromuscular and Neuroendocrinological Features Associated With ZC4H2-Related Arthrogryposis Multiplex Congenita in a Sicilian Family: A Case Report.
Frontiers in neurologyCharacterizing the molecular etiology of arthrogryposis multiplex congenita in patients with LGI4 mutations.
GliaUsing Telerehabilitation to Deliver a Home Exercise Program to Youth With Arthrogryposis: Single Cohort Pilot Study.
Journal of medical Internet researchInherited Defects of the ASC-1 Complex in Congenital Neuromuscular Diseases.
International journal of molecular sciencesNovel Approach to Improving Knee Range of Motion in Arthrogryposis with a New Working Classification.
Children (Basel, Switzerland)The One-Bone Forearm in Children: Surgical Technique and a Retrospective Review of Outcomes.
The Journal of hand surgeryInhibition of Maternal-to-Fetal Transfer of IgG Antibodies by FcRn Blockade in a Mouse Model of Arthrogryposis Multiplex Congenita.
Neurology(R) neuroimmunology & neuroinflammationA 7-year old female with arthrogryposis multiplex congenita, Duane retraction syndrome, and Marcus Gunn phenomenon due to a ZC4H2 gene mutation: a clinical presentation of the Wieacker-Wolff syndrome.
Ophthalmic geneticsBiallelic ASCC1 variants including a novel intronic variant result in expanded phenotypic spectrum of spinal muscular atrophy with congenital bone fractures 2 (SMABF2).
American journal of medical genetics. Part APhenotypic spectrum and genomics of undiagnosed arthrogryposis multiplex congenita.
Journal of medical geneticsMutation analysis and prenatal diagnosis of a family with congenital contractural arachnodactyly.
Molecular genetics & genomic medicineEmbryonic movement stimulates joint formation and development: Implications in arthrogryposis multiplex congenita.
BioEssays : news and reviews in molecular, cellular and developmental biologyPhysical therapy of temporomandibular disorder in a child with arthrogryposis multiplex congenita: A case report and literature review.
Cranio : the journal of craniomandibular practiceSingle-Stage Correction of Severe Rigid Ankle Equinus Deformity by Talectomy and Tibiocalcaneal Fusion in Adulthood: A Case Report.
Journal of orthopaedic case reportsFetal early motor neuron disruption and prenatal molecular diagnosis in a severe BICD2-opathy.
American journal of medical genetics. Part AA rare association of type 2 Duanes retraction syndrome with arthrogryposis multiplex congenita.
StrabismusGrowth-Friendly Spine Surgery in Arthrogryposis Multiplex Congenita.
The Journal of bone and joint surgery. American volumeAssistive and Rehabilitative Effects of the Playskin LiftTM Exoskeletal Garment on Reaching and Object Exploration in Children With Arthrogryposis.
The American journal of occupational therapy : official publication of the American Occupational Therapy AssociationConfirming the involvement of PIEZO2 in the etiology of Marden-Walker syndrome.
American journal of medical genetics. Part AAmyoplasia and distal arthrogryposis.
Orthopaedics & traumatology, surgery & research : OTSRConservative Treatment of Unicuspid Aortic Valve with Newly Diagnosed Type A Aortic Dissection.
Brazilian journal of cardiovascular surgeryOne-Stage Extension Shortening Osteotomy for Syndromic Camptodactyly.
Journal of clinical medicineA review of the orthopedic interventions and functional outcomes among a cohort of 114 children with arthrogryposis multiplex congenita.
Journal of pediatric rehabilitation medicinePrognostic significance of prenatal ultrasound in fetal arthrogryposis multiplex congenita.
Archives of gynecology and obstetricsArthrogryposis multiplex congenita with polymicrogyria and infantile encephalopathy caused by a novel GRIN1 variant.
Human genome variationNeurogenetic fetal akinesia and arthrogryposis: genetics, expanding genotype-phenotypes and functional genomics.
Journal of medical geneticsTotal joint replacement of the hip and knee in patients with arthrogryposis multiplex congenita: a report of six joints.
Archives of orthopaedic and trauma surgeryDe novo mutations of SCN1A are responsible for arthrogryposis broadening the SCN1A-related phenotypes.
Journal of medical geneticsPrenatal delineation of a distinct lethal fetal syndrome caused by a homozygous truncating KIDINS220 variant.
American journal of medical genetics. Part AA case of severe autosomal dominant spinal muscular atrophy with lower extremity predominance caused by a de novo BICD2 mutation.
Brain & developmentAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Artrogripose múltipla congênita.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Artrogripose múltipla congênita
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Management of Lower-Extremity Deformity in Arthrogryposis Multiplex Congentia: A Narrative Review.Medical science monitor : international medical journal of experimental and clinical research· 2026· PMID 41821200mais citado
- Arthrogryposis as a neuromuscular phenotype: lessons from PIEZO2 loss-of-function.
- CRISPR Activation Reveals the Spliceogenicity of an Intronic NEB Variant in Fetuses With Arthrogryposis Multiplex Congenita 6.
- Perinatal genetic diagnostic yield in a population of fetuses with the phenotype arthrogryposis multiplex congenita: a cohort study 2007-2021.
- Expanding the Phenotypic Spectrum of Syndromic Arthrogryposis Multiplex Congenita: Role of Biallelic Variants in COL25A1 Across Fetal and Pediatric Periods.
- Prenatal Diagnosis of Arthrogryposis Multiplex Congenita (AMC): Ultrasound and Genetic Findings in 69 Fetuses From 67 Unrelated Families.
- Growth arrest following posterior knee release in children with arthrogryposis multiplex congenita: a previously unreported complication.
- Milestone Attainment in Young Children With Arthrogryposis Multiplex Congenita: Developmental Profile and Associated Factors.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1037(Orphanet)
- MONDO:0015168(MONDO)
- GARD:777(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55785297(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
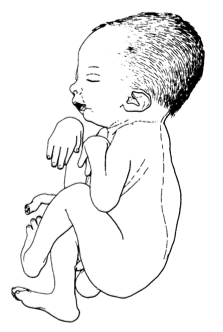
Artrogripose múltipla congênita
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov