A síndrome de artrogripose múltipla congênita tipo "face de assobiador" é uma forma raríssima de artrogripose múltipla congênita (uma condição caracterizada por múltiplas articulações rígidas e curvadas desde o nascimento). Ela se manifesta através de uma combinação de características, incluindo: * Rigidez e curvatura em várias articulações do corpo, o que limita os movimentos. * Boca pequena com formato que lembra um assobio, podendo causar dificuldades para se alimentar, engolir e falar. * Um rosto com uma expressão peculiar, geralmente com poucas expressões faciais. * Atraso grave no desenvolvimento. * Problemas no funcionamento do sistema nervoso central e autônomo, manifestados por: * Excesso de saliva. * Dificuldade para manter a temperatura corporal estável. * Crises epilépticas com espasmos musculares (convulsões mioclônicas). * Batimentos cardíacos lentos (bradicardia). * Ocasionalmente, a presença da sequência de Pierre Robin (uma condição que afeta o desenvolvimento da mandíbula e da boca). * Geralmente, leva à morte nos primeiros meses de vida. Sugere-se que a síndrome de artrogripose múltipla congênita tipo "face de assobiador" seja um conjunto de deformidades que surgem devido à falta de movimento do feto durante a gestação (acinesia fetal).
Introdução
O que você precisa saber de cara
A síndrome de artrogripose múltipla congênita tipo "face de assobiador" é uma forma raríssima de artrogripose múltipla congênita (uma condição caracterizada por múltiplas articulações rígidas e curvadas desde o nascimento). Ela se manifesta através de uma combinação de características, incluindo: * Rigidez e curvatura em várias articulações do corpo, o que limita os movimentos. * Boca pequena com formato que lembra um assobio, podendo causar dificuldades para se alimentar, engolir e falar. * Um rosto com uma expressão peculiar, geralmente com poucas expressões faciais. * Atraso grave no desenvolvimento. * Problemas no funcionamento do sistema nervoso central e autônomo, manifestados por: * Excesso de saliva. * Dificuldade para manter a temperatura corporal estável. * Crises epilépticas com espasmos musculares (convulsões mioclônicas). * Batimentos cardíacos lentos (bradicardia). * Ocasionalmente, a presença da sequência de Pierre Robin (uma condição que afeta o desenvolvimento da mandíbula e da boca). * Geralmente, leva à morte nos primeiros meses de vida. Sugere-se que a síndrome de artrogripose múltipla congênita tipo "face de assobiador" seja um conjunto de deformidades que surgem devido à falta de movimento do feto durante a gestação (acinesia fetal).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 13 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 32 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de artrogripose múltipla congênita-face em assobio
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome de artrogripose múltipla congênita-face em assobio
Centros para Síndrome de artrogripose múltipla congênita-face em assobio
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Genotype-Phenotype Landscape of NALCN and UNC80-Related Disorders.
The NALCN channelosome regulates the resting membrane potential through sodium leak currents, influencing cellular excitability. Genetic variants in NALCN and UNC80, a subunit of the NALCN channelosome, cause ultra-rare and severe neurodevelopmental disorders. Autosomal dominant congenital contractures of the limbs and face, hypotonia, and developmental delay (CLIFAHDD) syndrome is associated with gain-of-function (GOF) variants in NALCN. Infantile hypotonia with psychomotor retardation and characteristic facies (IHPRF) 1 syndrome is associated with biallelic variants in NALCN and IHPRF 2 syndrome with biallelic variants in UNC80, both resulting in a loss-of-function (LOF). This study aims to expand the phenotypes associated with these syndromes, exploring potential genotype-phenotype associations. This is a cross-sectional study including patients with pathogenic or likely pathogenic variants in NALCN and UNC80. Phenotypes were evaluated through a structured interview, questionnaires, and review of medical records. Associations between variants, clinical features, and syndromes were analyzed. Fifty-one patients were included (34 with CLIFAHDD, 9 with IHPRF 1, 8 with IHPRF 2; 3 months-27 years; 37.3% female). All exhibited neurodevelopmental delay, more severe in patients with LOF variants (p = 0.019). Neurodevelopmental regression was observed in 29.4% of patients with CLIFAHDD syndrome, associated with the onset of ataxia (70%). Patients with CLIFAHDD had more severe respiratory symptoms at birth (11.7% orotracheal intubation). Distal arthrogryposis (76.5%), episodic ataxia (41.2% of ambulatory patients), and paroxysmal dystonia (11.7%) were exclusively diagnosed in patients with CLIFAHDD. Patients with LOF variants presented more frequently with failure to thrive (88.2%, p = 0.001), central sleep apnea (CSA, 64.7%, p < 0.001), and epilepsy (70.6%, p < 0.001). Epilepsy was associated with more severe cognitive delays (p = 0.016) and was refractory in 58.8% of patients. Earlier seizure onset was associated with refractory epilepsy (p = 0.014). Patients with CLIFAHDD and premature death, epilepsy, or paroxysmal dystonia carried variants within NALCN pore domains. This study provides an in-depth clinical characterization of NALCN-related and UNC80-related disorders. Distal arthrogryposis, episodic ataxia, and paroxysmal dystonia were diagnosed in patients with CLIFAHDD while failure to thrive, CSA, and epilepsy were associated with LOF variants. We suggest potential genotype-phenotype associations, formulating hypotheses for validation in future studies with larger cohorts.
Unsafe Care and Fake News in Freeman-Burian Syndrome.
Freeman-Burian syndrome is a rare craniofacial syndrome surrounded by fake news. This situation shows the strong connection between the quality of a literature search and clinical reasoning displayed in patient care, especially in care of patients with rare conditions.
New presentation of CLIFAHDD syndrome with a novel variant in NALCN gene: A report of a rare case.
Congenital Contractures of Limbs and Face, Hypotonia, and Developmental Delay (CLIFAHDD) syndrome is a recently described type of distal arthrogryposis which unlike other subtypes is associated with developmental delay and various neurologic presentation. Epilepsy and ataxia have been reported. We add paroxysmal dyskinesia to the clinical spectrum. Understanding the molecular mechanism can help developing targeted therapy in future. This study resulted in identification of a novel variant in NALCN gene leading to autosomal dominant CLIFAHDD syndrome. Our patient presented with a form of nonepileptic paroxysmal dyskinesia. This is a new phenotype that has not been described previously.
Neurodevelopmental outcomes in a cohort of children with congenital Zika syndrome at 12 and 24 months of age.
Early child development is a critical stage of life that influences social, educational and health outcomes worldwide. A few years after Zika epidemic, families of children born with congenital Zika syndrome (CZS) continue to face uncertainties when it comes to the development of their children. The present study sought to analyse the developmental trajectories of a subset of children born with CZS in the first 24 months of life. Thirty-five children with CZS were assessed with the Bayley-III Scales at 12 and 24 months of age from November 2016 to December 2018 in a rehabilitation centre in Brazil. Inclusion criteria included children with established diagnosis of CZS. Exclusion criteria included the presence of arthrogryposis, prematurity, irregular follow-up, clinical complications or other causes of microcephaly. Children born with CZS who evolved with cerebral palsy (CP) were classified according to the Gross Motor Function Classification System (GMFCS) at 2 years of age. At 12 months of age mean composite scores on the Bayley cognitive, communication and motor scores were 57.71 (SD 7.11), 57.94 (SD 14.34) and 49.26 (7.20), respectively. At 24 months of age, composite scores were 57.43 (SD 7.11), 53.60 (SD 12.29) and 48.83 (7.76). In addition, 31 (88.57%) out of 34 children diagnosed with CP were classified as GMFCS levels IV and V. Zika virus congenital infection is a risk factor for functional impairments across all developmental domains having a direct and substantial negative impact in early child development.
Oculoplastic surgery, diagnosis, and other matters in Freeman-Burian syndrome.
Publicações recentes
Oculoplastic surgery, diagnosis, and other matters in Freeman-Burian syndrome.
📖 RevisãoNALCN channelopathies: Distinguishing gain-of-function and loss-of-function mutations.
Bilateral patellar tendon-bearing Symes-type prostheses in a severe case of Freeman-Sheldon syndrome in a 21-year-old woman presenting with uncorrectable equinovarus.
🥈 ObservacionalPreliminary experience with delayed non-operative therapy of multiple hand and wrist contractures in a woman with Freeman-Sheldon syndrome, at ages 24 and 28 years.
🥇 Revisão sistemáticaMicrodeletion 3q syndrome.
📚 EuropePMCmostrando 24
Genotype-Phenotype Landscape of NALCN and UNC80-Related Disorders.
NeurologyUnsafe Care and Fake News in Freeman-Burian Syndrome.
Clinical case reportsNew presentation of CLIFAHDD syndrome with a novel variant in NALCN gene: A report of a rare case.
Clinical case reportsNeurodevelopmental outcomes in a cohort of children with congenital Zika syndrome at 12 and 24 months of age.
Child: care, health and developmentOculoplastic surgery, diagnosis, and other matters in Freeman-Burian syndrome.
Ophthalmic geneticsA New Aberration in the VPS33B Gene Leads to Full-Symptom ARCS1.
The American journal of case reportsA rare association of type 2 Duanes retraction syndrome with arthrogryposis multiplex congenita.
StrabismusFurther delineation of MYO18B-related autosomal recessive Klippel-Feil syndrome with myopathy and facial dysmorphism.
American journal of medical genetics. Part AIdentification and Recent Approaches for Evaluation and Management of Rehabilitation Concerns for Patients with Freeman-Burian Syndrome: Principles for Global Treatment.
Journal of pediatric geneticsA Case of Severe Early-Onset Neuropathy Caused by a Compound Heterozygous Deletion of the PMP22 Gene: Clinical and Neurographic Aspects.
NeuropediatricsRevisiting the Many Names of Freeman-Sheldon Syndrome.
The Journal of craniofacial surgeryImaging findings in congenital Zika virus infection syndrome: an update.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryInterstitial deletion 5p14.1-p15.2 and 5q14.3-q23.2 in a patient with clubfoot, blepharophimosis, arthrogryposis, and multiple congenital abnormalities.
American journal of medical genetics. Part AFindings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
Systematic reviewsNovel NALCN variant: altered respiratory and circadian rhythm, anesthetic sensitivity.
Annals of clinical and translational neurologyNALCN channelopathies: Distinguishing gain-of-function and loss-of-function mutations.
NeurologyMolecularly proven mosaicism in phenotypically normal parent of a girl with Freeman-Sheldon Syndrome caused by a pathogenic MYH3 mutation.
American journal of medical genetics. Part ADeficiency in the mRNA export mediator Gle1 impairs Schwann cell development in the zebrafish embryo.
NeuroscienceFreeman-Sheldon syndrome in a 29-year-old woman presenting with rare and previously undescribed features.
BMJ case reportsDevelopmental myosins: expression patterns and functional significance.
Skeletal muscleBilateral patellar tendon-bearing Symes-type prostheses in a severe case of Freeman-Sheldon syndrome in a 21-year-old woman presenting with uncorrectable equinovarus.
BMJ case reportsPreliminary experience with delayed non-operative therapy of multiple hand and wrist contractures in a woman with Freeman-Sheldon syndrome, at ages 24 and 28 years.
BMJ case reports[Ross syndrome presenting with asymptomatic tonic pupils].
Rinsho shinkeigaku = Clinical neurologyDe novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de artrogripose múltipla congênita-face em assobio.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de artrogripose múltipla congênita-face em assobio
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Genotype-Phenotype Landscape of NALCN and UNC80-Related Disorders.
- Unsafe Care and Fake News in Freeman-Burian Syndrome.
- New presentation of CLIFAHDD syndrome with a novel variant in NALCN gene: A report of a rare case.
- Neurodevelopmental outcomes in a cohort of children with congenital Zika syndrome at 12 and 24 months of age.
- Oculoplastic surgery, diagnosis, and other matters in Freeman-Burian syndrome.
- NALCN channelopathies: Distinguishing gain-of-function and loss-of-function mutations.
- Bilateral patellar tendon-bearing Symes-type prostheses in a severe case of Freeman-Sheldon syndrome in a 21-year-old woman presenting with uncorrectable equinovarus.
- Preliminary experience with delayed non-operative therapy of multiple hand and wrist contractures in a woman with Freeman-Sheldon syndrome, at ages 24 and 28 years.
- Microdeletion 3q syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1150(Orphanet)
- OMIM OMIM:208155(OMIM)
- MONDO:0008825(MONDO)
- GARD:792(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q55781669(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
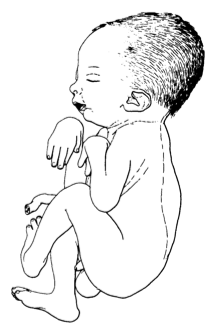
Síndrome de artrogripose múltipla congênita-face em assobio
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata