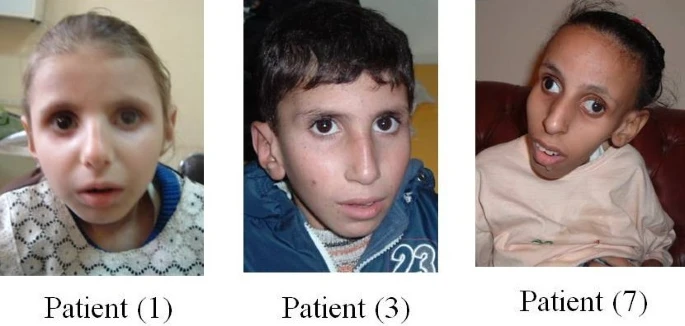
A Síndrome Cerebro-Óculo-Facio-Esquelética (COFS) é uma doença genética rara que pertence a um grupo de doenças que afetam o reparo do DNA (o "manual de instruções" das nossas células). Ela se caracteriza por causar problemas graves nos sentidos (como audição e visão) e no sistema nervoso.
Introdução
O que você precisa saber de cara
A Síndrome Cerebro-Óculo-Facio-Esquelética (COFS) é uma doença genética rara que pertence a um grupo de doenças que afetam o reparo do DNA (o "manual de instruções" das nossas células). Ela se caracteriza por causar problemas graves nos sentidos (como audição e visão) e no sistema nervoso.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 40 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 122 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Single-stranded structure-specific DNA endonuclease involved in DNA excision repair (PubMed:32522879, PubMed:32821917, PubMed:7651464, PubMed:8078765, PubMed:8090225, PubMed:8206890). Makes the 3'incision in DNA nucleotide excision repair (NER) (PubMed:32522879, PubMed:32821917, PubMed:8078765, PubMed:8090225). Binds and bends DNA repair bubble substrate and breaks base stacking at the single-strand/double-strand DNA junction of the DNA bubble (PubMed:32522879). Plays a role in base excision rep
NucleusChromosome
Xeroderma pigmentosum complementation group G
An autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. The skin develops marked freckling and other pigmentation abnormalities. Some XP-G patients present features of Cockayne syndrome, cachectic dwarfism, pigmentary retinopathy, ataxia, decreased nerve conduction velocities. The phenotype combining xeroderma pigmentosum and Cockayne syndrome traits is referred to as XP-CS complex.
ATP-dependent 5'-3' DNA helicase (PubMed:31253769, PubMed:8413672, PubMed:9771713). Component of the general transcription and DNA repair factor IIH (TFIIH) core complex, not absolutely essential for minimal transcription in vitro (PubMed:10024882, PubMed:17466626, PubMed:9771713). Required for transcription-coupled nucleotide excision repair (NER) of damaged DNA; recognizes damaged bases (PubMed:17466626, PubMed:23352696, PubMed:9771713). Sequestered in chromatin on UV-damaged DNA (PubMed:23352
NucleusCytoplasm, cytoskeleton, spindle
Xeroderma pigmentosum complementation group D
An autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. The skin develops marked freckling and other pigmentation abnormalities. Some XP-D patients present features of Cockayne syndrome, including cachectic dwarfism, pigmentary retinopathy, ataxia, decreased nerve conduction velocities. The phenotype combining xeroderma pigmentosum and Cockayne syndrome traits is referred to as XP-CS complex.
Essential factor involved in transcription-coupled nucleotide excision repair (TC-NER), a process during which RNA polymerase II-blocking lesions are rapidly removed from the transcribed strand of active genes (PubMed:16246722, PubMed:20541997, PubMed:22483866, PubMed:26620705, PubMed:32355176, PubMed:34526721, PubMed:38316879, PubMed:38600235, PubMed:38600236). Plays a central role in the initiation of the TC-NER process: specifically recognizes and binds RNA polymerase II stalled at a lesion,
NucleusChromosome
Cockayne syndrome B
A rare disorder characterized by cutaneous sensitivity to sunlight, abnormal and slow growth, cachectic dwarfism, progeroid appearance, progressive pigmentary retinopathy and sensorineural deafness. There is delayed neural development and severe progressive neurologic degeneration resulting in intellectual disability. Two clinical forms are recognized: in the classical form or Cockayne syndrome type 1, the symptoms are progressive and typically become apparent within the first few years or life; the less common Cockayne syndrome type 2 is characterized by more severe symptoms that manifest prenatally. Cockayne syndrome shows some overlap with certain forms of xeroderma pigmentosum. Unlike xeroderma pigmentosum, patients with Cockayne syndrome do not manifest increased freckling and other pigmentation abnormalities in the skin and have no significant increase in skin cancer.
Non-catalytic component of a structure-specific DNA repair endonuclease responsible for the 5'-incision during DNA repair. Responsible, in conjunction with SLX4, for the first step in the repair of interstrand cross-links (ICL). Participates in the processing of anaphase bridge-generating DNA structures, which consist in incompletely processed DNA lesions arising during S or G2 phase, and can result in cytokinesis failure. Also required for homology-directed repair (HDR) of DNA double-strand bre
NucleusCytoplasm
Cerebro-oculo-facio-skeletal syndrome 4
A disorder of prenatal onset characterized by microcephaly, congenital cataracts, facial dysmorphism, neurogenic arthrogryposis, growth failure and severe psychomotor retardation. COFS is considered to be part of the nucleotide-excision repair disorders spectrum that include also xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome.
Variantes genéticas (ClinVar)
1.099 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
34 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome COFS
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
DNA repair-related heritable photosensitivity syndromes: Mutation landscape in a multiethnic cohort of 17 multigenerational families with high degree of consanguinity.
Inherited photosensitivity syndromes are a heterogeneous group of genetic skin disorders with tremendous phenotypic variability, characterized by photosensitivity and defective DNA repair, especially nucleotide excision repair. A cohort of 17 Iranian families with heritable photosensitivity syndromes was evaluated to identify their genetic defect. The patients' DNA was analyzed with either whole-exome sequencing or RNA sequencing (RNA-Seq). The interpretations of the genomic results were guided by genome-wide homozygosity mapping. Haplotype analysis was performed for cases with recurrent mutations. RNA-Seq, in addition to mutation detection, was also utilized to confirm the pathogenicity. Thirteen sequence variants, including six previously unreported pathogenic variants, were disclosed in 17 Iranian families, with XPC as the most common mutated gene in 10 families (59%). In one patient, RNA-Seq, as a first-tier diagnostic approach, revealed a non-canonical homozygous germline variant: XPC:c.413-9 T > A. The Sashimi plot showed skipping of exon 4 with dramatic XPC down-expression. Haplotype analysis of XPC:c.2251-1 G>C and XPC:1243 C>T in four families showed common haplotypes of 1.7 Mb and 2.6 Mb, respectively, denoting a founder effect. Lastly, two extremely rare cases were presented in this report: a homozygous UVSSA:c .1990 C>T was disclosed, and ERCC2-related cerebro-oculo-facio-skeletal (COFS) syndrome with an early childhood death. A direct comparison of our data with the results of previously reported cohorts demonstrates the international mutation landscape of DNA repair-related photosensitivity disorders, although population-specific differences were observed.
Prenatal findings of cataract and arthrogryposis: recurrence of cerebro-oculo-facio-skeletal syndrome and review of differential diagnosis.
Cerebro-oculo-facio-skeletal syndrome (COFS) is a severe and progressive neurologic condition characterized by prenatal onset of arthrogryposis, cataract, microcephaly and growth failure. The aim of this study was to present a case of recurrence of the COFS syndrome and to propose a differential diagnosis flow-chart in case of prenatal findings of arthrogryposis and cataract. We report a case of recurrence of COFS3 syndrome within the same family, with similar diagnostic features. In the first case the COFS syndrome remained undiagnosed, while in the second case, due to prenatal findings of arthrogryposis and cataract, genetic investigation focusing on responsible genes of COFS (ERCC5, ERCC6 and FKTN genes) was carried out. The fetus was found to be compound heterozygous for two different ERCC5 mutations, confirming the clinical suspect of COFS syndrome. A review of the literature on possible causative genes of prenatal cataract and arthrogryposis was performed and we present a flow-chart to guide differential diagnosis and possible genetic testing in case of these findings. COFS syndrome is a rare autosomic recessive condition. However, it can be suspected and diagnosed prenatally. The flow-chart illustrates a pathway to guide differential diagnosis according to the prenatal findings. Main syndromes, key testing and specific genes are included. Targeted molecular testing should be offered to the couple in order to reach a diagnosis and assess the recurrence risk for future pregnancies.
Cerebro-oculo-facio-skeletal syndrome caused by the homozygous pathogenic variant Gly47Arg in ERCC2.
DNA damage repair is a pivotal mechanism in life. The nucleotide excision repair pathway protects the cells against DNA damage and involves XPD, an ATP dependent helicase that is part of the multisubunit protein complex TFIIH. XPD is encoded by the excision repair cross-complementation group 2 gene (ERCC2). Only three patients with cerebro-oculo-facio-skeletal syndrome (COFS), caused by mutations in ERCC2, have been published so far. This report describes a boy with the homozygous amino acid change p.Gly47Arg in XPD. He presented with profound microcephaly, psychomotor retardation, failure to thrive, cutaneous photosensitivity, a bilateral hearing deficit and optic atrophy, thrombocytopenia, and recurrent episodes of pneumonia. We report the first homozygous occurrence of the pathogenic variant Gly47Arg in the ERCC2 gene. Occurring homozygous, this variant was associated with COFS syndrome, leading to early death of the patient at the age of 21 months.
COFS type 3 in an Indian family with antenatally detected arthrogryposis.
Fetal akinesia and contractures can be caused by mutations in various genes that lead to overlapping phenotypes with contractures, rocker bottom feet, cerebellar hypoplasia, ventriculomegaly, growth retardation, pulmonary hypoplasia, cystic hygroma and cleft palate in various combinations. Cerebro-oculo-facio-skeletal (COFS) syndrome is a condition resulting from defects in DNA repair pathway, and genes involved include ERCC1 (COFS), ERCC2 (XPD), ERCC5(XPG), and ERCC6 (CSB). It is a severe disorder presenting in fetal or neonatal period with microcephaly, arthrogryposis, prominent nose, and kyphoscoliosis, and leads to early death in childhood. We report a baby with antenatally identified arthrogryposis in which the homozygous pathogenic variant in exon 8 was identified in ERCC5 gene, by targeted next generation sequencing. This was predicted to cause premature chain termination in the protein. ERCC5 gene is mainly implicated in xeroderma pigmentosum, sometimes in COFS syndrome.
Prenatal diagnosis of cerebro-oculo-facio-skeletal syndrome: Report of three fetuses and review of the literature.
Cerebro-oculo-facio-skeletal syndrome (COFS) is a rare autosomal recessive neurodegenerative disease belonging to the family of DNA repair disorders, characterized by microcephaly, congenital cataracts, facial dysmorphism and arthrogryposis. Here, we describe the detailed morphological and microscopic phenotype of three fetuses from two families harboring ERCC5/XPG likely pathogenic variants, and review the five previously reported fetal cases. In addition to the classical features of COFS, the fetuses display thymus hyperplasia, splenomegaly and increased hematopoiesis. Microencephaly is present in the three fetuses with delayed development of the gyri, but normal microscopic anatomy at the supratentorial level. Microscopic anomalies reminiscent of pontocerebellar hypoplasia are present at the infratentorial level. In conclusion, COFS syndrome should be considered in fetuses when intrauterine growth retardation is associated with microcephaly, arthrogryposis and ocular anomalies. Further studies are needed to better understand XPG functions during human development.
Publicações recentes
DNA repair-related heritable photosensitivity syndromes: Mutation landscape in a multiethnic cohort of 17 multigenerational families with high degree of consanguinity.
🥉 Relato de casoPrenatal findings of cataract and arthrogryposis: recurrence of cerebro-oculo-facio-skeletal syndrome and review of differential diagnosis.
Cerebro-oculo-facio-skeletal syndrome caused by the homozygous pathogenic variant Gly47Arg in ERCC2.
COFS type 3 in an Indian family with antenatally detected arthrogryposis.
📚 EuropePMC15 artigos no totalmostrando 5
DNA repair-related heritable photosensitivity syndromes: Mutation landscape in a multiethnic cohort of 17 multigenerational families with high degree of consanguinity.
DNA repairPrenatal findings of cataract and arthrogryposis: recurrence of cerebro-oculo-facio-skeletal syndrome and review of differential diagnosis.
BMC medical genomicsCerebro-oculo-facio-skeletal syndrome caused by the homozygous pathogenic variant Gly47Arg in ERCC2.
American journal of medical genetics. Part ACOFS type 3 in an Indian family with antenatally detected arthrogryposis.
American journal of medical genetics. Part APrenatal diagnosis of cerebro-oculo-facio-skeletal syndrome: Report of three fetuses and review of the literature.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome COFS.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome COFS
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- DNA repair-related heritable photosensitivity syndromes: Mutation landscape in a multiethnic cohort of 17 multigenerational families with high degree of consanguinity.
- Prenatal findings of cataract and arthrogryposis: recurrence of cerebro-oculo-facio-skeletal syndrome and review of differential diagnosis.
- Cerebro-oculo-facio-skeletal syndrome caused by the homozygous pathogenic variant Gly47Arg in ERCC2.
- COFS type 3 in an Indian family with antenatally detected arthrogryposis.
- Prenatal diagnosis of cerebro-oculo-facio-skeletal syndrome: Report of three fetuses and review of the literature.
- Cockayne Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1466(Orphanet)
- MONDO:0008926(MONDO)
- GARD:6027(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q29014951(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome COFS
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata