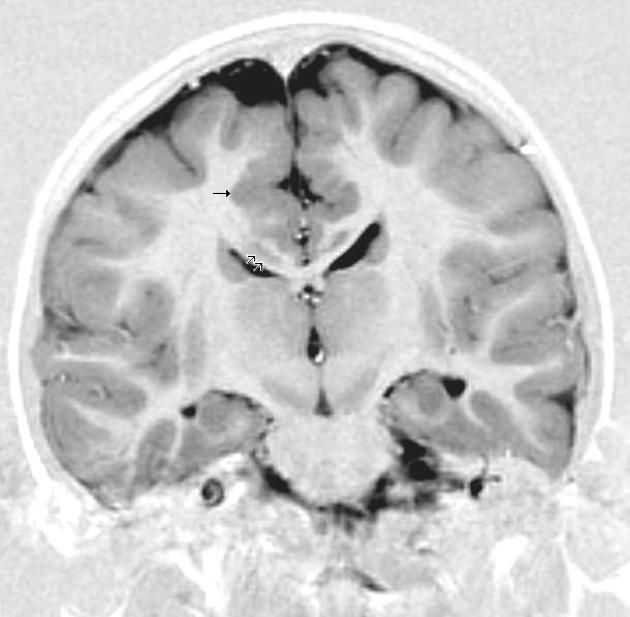
Malformação cerebral rara não sindrômica devido à migração anómala neuronal específica por aglomerados desorganizados de neurônios em locais atípicos, como a localização periventricular e subcortical. A extensão das lesões varia de nódulos confluentes únicos a bilaterais isolados. Os pacientes em idade pediátrica habitualmente apresentam graus variáveis de perturbação do desenvolvimento intelectual e epilepsia intratável, e as malformações específicas e/ou sistêmicas concomitantes são frequentes. Nas formas mais ligeiras ou no início das convulsões podem ocorrer apenas na idade adulta.
Introdução
O que você precisa saber de cara
Visão geral
A Heterotopia nodular hereditária é uma condição rara do desenvolvimento neurológico em que grupos de neurônios (células nervosas) não migram para a posição correta durante a formação do cérebro, formando nódulos na substância branca ou próximo aos ventrículos cerebrais. Essa alteração pode estar associada a anormalidades na estrutura do córtex cerebral e a uma variedade de manifestações clínicas que afetam diferentes sistemas do corpo. A prevalência exata da doença é desconhecida, e sua apresentação pode ocorrer em qualquer idade.[1][3]
Sinais e sintomas
Os sinais e sintomas da Heterotopia nodular hereditária são variados e podem incluir alterações neurológicas, como fraqueza muscular, anormalidades no eletroencefalograma (EEG) — incluindo atividade lenta focal, ondas de espícula focais temporais e ondas lentas agudas focais — e mioclonia de membro (movimentos musculares involuntários). Além disso, podem ocorrer manifestações em outros sistemas, como pé cavo, escoliose, hipermobilidade articular, luxação do ombro ou patelar, hérnia, estenose pilórica, refluxo gastroesofágico, pele fina, sangramento anormal, regurgitação aórtica, aneurisma da aorta, morfologia anormal da válvula cardíaca, anormalidades da testa, do forame magno e mielomeningocele.[1][3]
Causas genéticas
A Heterotopia nodular hereditária é causada por alterações (mutações) em genes que orientam a migração neuronal durante o desenvolvimento embrionário. Os padrões de herança podem ser autossômico dominante, autossômico recessivo ou ligado ao cromossomo X. Os genes associados à condição incluem: FLNA (Filamin-A), TMTC3 (Protein O-mannosyl-transferase TMTC3), ERMARD (Endoplasmic reticulum membrane-associated RNA degradation protein), MAP1B (Microtubule-associated protein 1B), ARFGEF2 (Brefeldin A-inhibited guanine nucleotide-exchange protein 2), NEDD4L (E3 ubiquitin-protein ligase NEDD4-like) e ARF1 (ADP-ribosylation factor 1).[1][4]
Diagnóstico
O diagnóstico da Heterotopia nodular hereditária é baseado na avaliação clínica, exames de imagem do sistema nervoso (como ressonância magnética) e confirmação genética. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para procedimentos diagnósticos, incluindo: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); dosagem de alfa-fetoproteína; e atendimento em reabilitação para doenças raras. Atualmente, há 12 tipos de testes genéticos disponíveis e 1.307 variantes registradas no ClinVar para essa condição.[1][4]
Tratamento e manejo
O tratamento da Heterotopia nodular hereditária é individualizado e focado no manejo dos sintomas apresentados por cada paciente. Pode incluir acompanhamento neurológico, fisioterapia, terapia ocupacional e suporte para complicações específicas, como escoliose, refluxo gastroesofágico ou alterações cardíacas. Não há medicamentos específicos aprovados para a condição, e o manejo deve ser realizado por uma equipe multidisciplinar. O SUS oferece atendimento em reabilitação para doenças raras como parte do suporte disponível.[1]
Prognóstico e qualidade de vida
O prognóstico da Heterotopia nodular hereditária varia amplamente, dependendo da gravidade das manifestações neurológicas e sistêmicas. Como a condição pode se apresentar em qualquer idade, o acompanhamento regular com especialistas é fundamental para monitorar complicações e ajustar o plano de cuidados. O suporte multidisciplinar e o acesso a reabilitação podem contribuir para melhorar a qualidade de vida dos pacientes e suas famílias.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Malformação cerebral rara não sindrômica devido à migração anómala neuronal específica por aglomerados desorganizados de neurônios em locais atípicos, como a localização periventricular e subcortical. A extensão das lesões varia de nódulos confluentes únicos a bilaterais isolados. Os pacientes em idade pediátrica habitualmente apresentam graus variáveis de perturbação do desenvolvimento intelectual e epilepsia intratável, e as malformações específicas e/ou sistêmicas concomitantes são frequentes. Nas formas mais ligeiras ou no início das convulsões podem ocorrer apenas na idade adulta.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Heterotopia nodular hereditária é uma condição rara do desenvolvimento neurológico em que grupos de neurônios (células nervosas) não migram para a posição correta durante a formação do cérebro, formando nódulos na substância branca ou próximo aos ventrículos cerebrais. Essa alteração pode estar associada a anormalidades na estrutura do córtex cerebral e a uma variedade de manifestações clínicas que afetam diferentes sistemas do corpo. A prevalência exata da doença é desconhecida, e sua apresentação pode ocorrer em qualquer idade.[1][3]
Sinais e sintomas
Os sinais e sintomas da Heterotopia nodular hereditária são variados e podem incluir alterações neurológicas, como fraqueza muscular, anormalidades no eletroencefalograma (EEG) — incluindo atividade lenta focal, ondas de espícula focais temporais e ondas lentas agudas focais — e mioclonia de membro (movimentos musculares involuntários). Além disso, podem ocorrer manifestações em outros sistemas, como pé cavo, escoliose, hipermobilidade articular, luxação do ombro ou patelar, hérnia, estenose pilórica, refluxo gastroesofágico, pele fina, sangramento anormal, regurgitação aórtica, aneurisma da aorta, morfologia anormal da válvula cardíaca, anormalidades da testa, do forame magno e mielomeningocele.[1][3]
Causas genéticas
A Heterotopia nodular hereditária é causada por alterações (mutações) em genes que orientam a migração neuronal durante o desenvolvimento embrionário. Os padrões de herança podem ser autossômico dominante, autossômico recessivo ou ligado ao cromossomo X. Os genes associados à condição incluem: FLNA (Filamin-A), TMTC3 (Protein O-mannosyl-transferase TMTC3), ERMARD (Endoplasmic reticulum membrane-associated RNA degradation protein), MAP1B (Microtubule-associated protein 1B), ARFGEF2 (Brefeldin A-inhibited guanine nucleotide-exchange protein 2), NEDD4L (E3 ubiquitin-protein ligase NEDD4-like) e ARF1 (ADP-ribosylation factor 1).[1][4]
Diagnóstico
O diagnóstico da Heterotopia nodular hereditária é baseado na avaliação clínica, exames de imagem do sistema nervoso (como ressonância magnética) e confirmação genética. No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para procedimentos diagnósticos, incluindo: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); dosagem de alfa-fetoproteína; e atendimento em reabilitação para doenças raras. Atualmente, há 12 tipos de testes genéticos disponíveis e 1.307 variantes registradas no ClinVar para essa condição.[1][4]
Tratamento e manejo
O tratamento da Heterotopia nodular hereditária é individualizado e focado no manejo dos sintomas apresentados por cada paciente. Pode incluir acompanhamento neurológico, fisioterapia, terapia ocupacional e suporte para complicações específicas, como escoliose, refluxo gastroesofágico ou alterações cardíacas. Não há medicamentos específicos aprovados para a condição, e o manejo deve ser realizado por uma equipe multidisciplinar. O SUS oferece atendimento em reabilitação para doenças raras como parte do suporte disponível.[1]
Prognóstico e qualidade de vida
O prognóstico da Heterotopia nodular hereditária varia amplamente, dependendo da gravidade das manifestações neurológicas e sistêmicas. Como a condição pode se apresentar em qualquer idade, o acompanhamento regular com especialistas é fundamental para monitorar complicações e ajustar o plano de cuidados. O suporte multidisciplinar e o acesso a reabilitação podem contribuir para melhorar a qualidade de vida dos pacientes e suas famílias.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 48 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 145 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
7 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, X-linked dominant.
Promotes orthogonal branching of actin filaments and links actin filaments to membrane glycoproteins. Anchors various transmembrane proteins to the actin cytoskeleton and serves as a scaffold for a wide range of cytoplasmic signaling proteins. Interaction with FLNB may allow neuroblast migration from the ventricular zone into the cortical plate. Tethers cell surface-localized furin, modulates its rate of internalization and directs its intracellular trafficking (By similarity). Involved in cilio
Cytoplasm, cell cortexCytoplasm, cytoskeletonPerikaryonCell projection, growth coneCell projection, podosome
Periventricular nodular heterotopia 1
A developmental disorder characterized by the presence of periventricular nodules of cerebral gray matter, resulting from a failure of neurons to migrate normally from the lateral ventricular proliferative zone, where they are formed, to the cerebral cortex. PVNH1 is an X-linked dominant form. Heterozygous females have normal intelligence but suffer from seizures and various manifestations outside the central nervous system, especially related to the vascular system. Hemizygous affected males die in the prenatal or perinatal period.
Transfers mannosyl residues to the hydroxyl group of serine or threonine residues. The 4 members of the TMTC family are O-mannosyl-transferases dedicated primarily to the cadherin superfamily, each member seems to have a distinct role in decorating the cadherin domains with O-linked mannose glycans at specific regions. Also acts as O-mannosyl-transferase on other proteins such as PDIA3 (PubMed:28973932). Involved in the positive regulation of proteasomal protein degradation in the endoplasmic re
MembraneEndoplasmic reticulum
Lissencephaly 8
A form of lissencephaly, a disorder of cortical development characterized by agyria or pachygyria and disorganization of the clear neuronal lamination of normal six-layered cortex. LIS8 patients manifest delayed psychomotor development, intellectual disability with poor or absent speech, early-onset refractory seizures, hypotonia, cortical gyral abnormalities, and hypoplasia of the corpus callosum, brainstem and cerebellum. LIS8 inheritance is autosomal recessive.
May play a role in neuronal migration during embryonic development
Endoplasmic reticulum membrane
Periventricular nodular heterotopia 6
A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH6 results in delayed psychomotor development, delayed speech, strabismus, and onset of seizures with hypsarrhythmia in early infancy.
Facilitates tyrosination of alpha-tubulin in neuronal microtubules (By similarity). Phosphorylated MAP1B is required for proper microtubule dynamics and plays a role in the cytoskeletal changes that accompany neuronal differentiation and neurite extension (PubMed:33268592). Possibly MAP1B binds to at least two tubulin subunits in the polymer, and this bridging of subunits might be involved in nucleating microtubule polymerization and in stabilizing microtubules. Acts as a positive cofactor in DA
Cytoplasm, cytoskeletonCytoplasmSynapseCell projection, dendritic spine
Periventricular nodular heterotopia 9
A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH9 is an autosomal dominant disorder with incomplete penetrance, characterized by impaired intellectual development, cognitive defects, learning disabilities, and behavior abnormalities. Some patients develop seizures.
Promotes guanine-nucleotide exchange on ARF1 and ARF3 and to a lower extent on ARF5 and ARF6. Promotes the activation of ARF1/ARF5/ARF6 through replacement of GDP with GTP. Involved in the regulation of Golgi vesicular transport. Required for the integrity of the endosomal compartment. Involved in trafficking from the trans-Golgi network (TGN) to endosomes and is required for membrane association of the AP-1 complex and GGA1. Seems to be involved in recycling of the transferrin receptor from rec
CytoplasmMembraneGolgi apparatusCytoplasm, perinuclear regionGolgi apparatus, trans-Golgi networkEndosomeCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, dendriteCytoplasmic vesicleSynapseCytoplasm, cytoskeleton
Periventricular nodular heterotopia 2
A developmental disorder characterized by the presence of periventricular nodules of cerebral gray matter, resulting from a failure of neurons to migrate normally from the lateral ventricular proliferative zone, where they are formed, to the cerebral cortex. PVNH2 is an autosomal recessive form characterized by microcephaly (small brain), severe developmental delay and recurrent infections. No anomalies extrinsic to the central nervous system, such as dysmorphic features or grossly abnormal endocrine or other conditions, are associated with PVNH2.
E3 ubiquitin-protein ligase that mediates the polyubiquitination of lysine and cysteine residues on target proteins and is thereby implicated in the regulation of various signaling pathways including autophagy, innate immunity or DNA repair (PubMed:20064473, PubMed:31959741, PubMed:33608556). Inhibits TGF-beta signaling by triggering SMAD2 and TGFBR1 ubiquitination and proteasome-dependent degradation (PubMed:15496141). Downregulates autophagy and cell growth by ubiquitinating and reducing cellu
CytoplasmGolgi apparatusEndosome, multivesicular body
Periventricular nodular heterotopia 7
A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH7 is an autosomal dominant disease characterized by delayed psychomotor development, intellectual disability, and seizures in some patients. Additional features include cleft palate and toe syndactyly.
Small GTPase involved in protein trafficking between different compartments (PubMed:8253837). Modulates vesicle budding and uncoating within the Golgi complex (PubMed:8253837). In its GTP-bound form, triggers the recruitment of coatomer proteins to the Golgi membrane (PubMed:8253837). The hydrolysis of ARF1-bound GTP, which is mediated by ARFGAPs proteins, is required for dissociation of coat proteins from Golgi membranes and vesicles (PubMed:8253837). The GTP-bound form interacts with PICK1 to
Golgi apparatus membraneSynapse, synaptosomePostsynaptic density
Periventricular nodular heterotopia 8
A form of periventricular nodular heterotopia, a disorder resulting from a defect in the pattern of neuronal migration in which ectopic collections of neurons lie along the lateral ventricles of the brain or just beneath, contiguously or in isolated patches. PVNH8 is an autosomal dominant disease characterized by developmental disabilities, speech delay, seizures and attention deficit-hyperactivity disorder.
Variantes genéticas (ClinVar)
1.307 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
25 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Heterotopia nodular hereditária
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Publicações recentes
X-linked pyruvate dehydrogenase complex deficiency due to a novel PDHA1 variant associated with structural brain abnormalities in a fetus.
Characterization of nodular neuronal heterotopia in children.
Neuronal migration disorders: a contribution of modern neuroimaging to the etiologic diagnosis of epilepsy.
📚 EuropePMC1 artigos no totalmostrando 1
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Heterotopia nodular hereditária.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Heterotopia nodular hereditária
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- X-linked pyruvate dehydrogenase complex deficiency due to a novel PDHA1 variant associated with structural brain abnormalities in a fetus.
- Characterization of nodular neuronal heterotopia in children.
- Neuronal migration disorders: a contribution of modern neuroimaging to the etiologic diagnosis of epilepsy.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2149(Orphanet)
- MONDO:0016292(MONDO)
- GARD:16586(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q25397988(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Heterotopia nodular hereditária
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata