A síndrome oral-facio-digital tipo 8 é caracterizada por lobulação da língua, hipoplasia da epiglote, fenda mediana do lábio superior, ponta nasal larga ou bífida, hipertelorismo ou telecanto, polidactilia pré-axial e pós-axial bilateral, tíbias e/ou rádios anormais, duplicação dos hálux, baixa estatura e déficit intelectual leve.
Introdução
O que você precisa saber de cara
A síndrome oral-facio-digital tipo 8 é caracterizada por lobulação da língua, hipoplasia da epiglote, fenda mediana do lábio superior, ponta nasal larga ou bífida, hipertelorismo ou telecanto, polidactilia pré-axial e pós-axial bilateral, tíbias e/ou rádios anormais, duplicação dos hálux, baixa estatura e déficit intelectual leve.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 5 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 18 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome oro-facio-digital tipo 8
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 6 publicações de um total de 50
Reduced cone photoreceptor function and subtle systemic manifestations in two siblings with loss of SCLT1.
The sodium channel and clathrin linker 1 gene (SCLT1) has been involved in the pathogenesis of various ciliopathy disorders such as Bardet-Biedl syndrome, orofaciodigital syndrome type IX, and Senior-Løken syndrome. Detailed exams are warranted to outline all clinical features. Here, we present a family with a milder phenotype of SCLT1-related disease. Comprehensive eye examination including fundus images, OCT, color vision, visual fields and electroretinography were performed. Affected individuals were assessed by a pediatrician and a medical geneticist for systemic features of ciliopathy. Investigations included echocardiography, abdominal ultrasonography, blood work-up for diabetes, liver and kidney function. Genetic testing included NGS retinal dystrophy panel, segregation analysis and transcriptome sequencing. Two male children, age 10 and 8 years, were affected with attention deficit hyperactivity disorder (ADHD), obesity and mild photophobia. The ophthalmic exam revealed reduced best-corrected visual acuity (BCVA), strabismus, hyperopia, astigmatism and moderate red-green defects. Milder changes suggesting photoreceptors disease were found on retinal imaging. Electroretinogram confirmed cone photoreceptors dysfunction. Genetic testing revealed a homozygous likely pathogenic, splice-site variant in SCLT1 gene NM_144643.3: c.1439 + 1del in the proband and in the affected brother. The unaffected parents were heterozygous for the SCLT1 variant. Transcriptome sequencing showed retention of intron 16 in the proband. In this report, we highlight the importance of further extensive diagnostics in patients with unexplained reduced vision, strabismus, refractive errors and ADHD spectrum disorders. SCLT1-related retinal degeneration is very rare and isolated reduced function of cone photoreceptors has not previously been observed.
Whole-Exon Sequencing and Correlation Analysis of a 14-Month-Old Girl With Orofaciodigital Syndrome.
Orofaciodigital syndrome type 1 (OFDS1) is a genetic disorder characterized by specific oral, facial, and limb malformations. A 14-month-old girl with congenital cleft palate, lower lip midline cleft, and digital anomalies admitted to our hospital was preliminarily diagnosed with OFDS1. Genetic analysis revealed that she carried a heterozygous variant of OFD1 at locus Xp22.2 on the X chromosome. Herein, we present the specific phenotype and genotype and the treatment modalities for this patient and references for diagnosing and treating OFDS.
Clinical and Descriptive Study of Orofacial Clefts in Colombia: 2069 Patients From Operation Smile Foundation.
To describe the population of patients with cleft lip and/or palate (CL/P) in terms of cleft phenotypes, gender, age, ethnic group, family history, clinical presentation (syndromic vs nonsyndromic), some environmental and behavioral factors, and some clinical features. Descriptive retrospective study. Patients attending the genetics counseling practice in Operation Smile Foundation, Bogotá, Colombia, for over 8 years. No screening was conducted. All patients requiring clinical genetics assessment in Operation Smile Foundation were included in the study. Left cleft lip and palate (CLP) and nonsyndromic forms were the most frequent types of malformations in this population. Psychomotor retardation and heart disease were the most frequent comorbidities in these patients. A low proportion of mothers exposed to passive smoking during pregnancy was observed and low birth weight accounted for an important number of cases. Aarskog, velocardiofacial, and orofaciodigital syndromes were the most frequent syndromic forms of CLP in this population. In this study, the most frequent type of CL/P was the nonsyndromic complete left CLP. Aarskog, velocardiofacial, and orofaciodigital syndromes were the most frequent syndromic forms of CL/P in this population.
Novel Surgical Technique for Correction of Incomplete Median Cleft Lip Deformity in Oral-Facial-Digital Syndrome Type II.
Oral-facial-digital syndromes (OFDSs) represent a heterogenous group of embryonic development disorders characterized by malformations of the face, oral cavity, and extremities. Oral-facial-digital syndrome type II is an autosomal recessive disease characterized by median cleft lip, gingival frenula, cleft lobulated tongue, and polydactyly. There are few reports on surgical techniques for correction of incomplete median cleft lip. Here we describe a novel surgical method that we used to correct an incomplete median cleft lip in a 5-year-old girl with oral-facial-digital syndrome type II. She had previously undergone surgery for congenital heart disease, oral anomalies, and polydactyly. Cheiloplasty was performed at 5 years and 8 months using a surgical approach that focused on repair of the median tubercle using lateral labial elements. A reasonably good Cupid's bow and median tubercle were achieved. Our technique for surgical correction of moderate incomplete median cleft lip provides adequate philtral height, vermillion fullness, and a good-shaped Cupid's bow.
Prenatal findings in oral-facial-digital syndrome type VI: Report of three cases and literature review.
Publicações recentes
RNA Sequencing for Rare Disease Diagnosis in a South African Family: A Novel Exon Elongation Event in OFD1.
The luminal ring protein C2CD3 acts as a radial in-to-out organizer of the distal centriole and appendages.
Expanding the phenotype associated with biallelic SCNM1 variants.
Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
The Luminal Ring Protein C2CD3 Acts as a Radial In-to-Out Organizer of the Distal Centriole and Appendages.
📚 EuropePMC86 artigos no totalmostrando 6
Reduced cone photoreceptor function and subtle systemic manifestations in two siblings with loss of SCLT1.
Ophthalmic geneticsWhole-Exon Sequencing and Correlation Analysis of a 14-Month-Old Girl With Orofaciodigital Syndrome.
The Journal of craniofacial surgeryNovel Surgical Technique for Correction of Incomplete Median Cleft Lip Deformity in Oral-Facial-Digital Syndrome Type II.
The Journal of craniofacial surgeryClinical and Descriptive Study of Orofacial Clefts in Colombia: 2069 Patients From Operation Smile Foundation.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationPrenatal findings in oral-facial-digital syndrome type VI: Report of three cases and literature review.
Prenatal diagnosisExpanding the allelic disorders linked to TCTN1 to include Varadi syndrome (Orofaciodigital syndrome type VI).
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome oro-facio-digital tipo 8.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome oro-facio-digital tipo 8
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Reduced cone photoreceptor function and subtle systemic manifestations in two siblings with loss of SCLT1.
- Whole-Exon Sequencing and Correlation Analysis of a 14-Month-Old Girl With Orofaciodigital Syndrome.
- Clinical and Descriptive Study of Orofacial Clefts in Colombia: 2069 Patients From Operation Smile Foundation.The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial Association· 2022· PMID 33736479mais citado
- Novel Surgical Technique for Correction of Incomplete Median Cleft Lip Deformity in Oral-Facial-Digital Syndrome Type II.
- Prenatal findings in oral-facial-digital syndrome type VI: Report of three cases and literature review.
- RNA Sequencing for Rare Disease Diagnosis in a South African Family: A Novel Exon Elongation Event in OFD1.
- The luminal ring protein C2CD3 acts as a radial in-to-out organizer of the distal centriole and appendages.
- Expanding the phenotype associated with biallelic SCNM1 variants.
- Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
- The Luminal Ring Protein C2CD3 Acts as a Radial In-to-Out Organizer of the Distal Centriole and Appendages.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2755(Orphanet)
- OMIM OMIM:300484(OMIM)
- MONDO:0010336(MONDO)
- GARD:4060(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q21154047(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
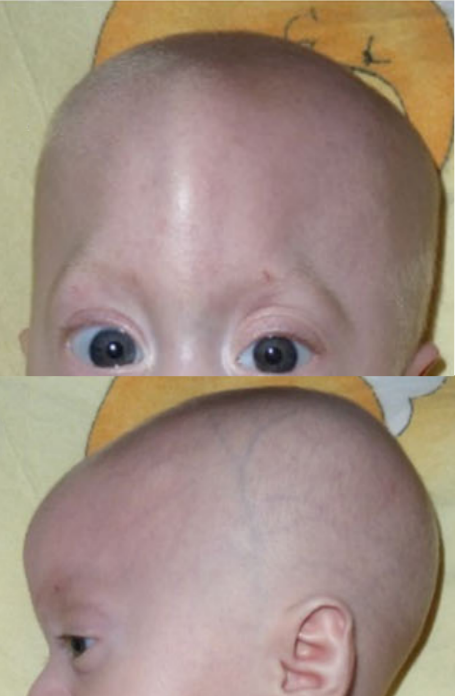
Síndrome oro-facio-digital tipo 8
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata