A paraplegia espástica autossômica recessiva tipo 15 é uma forma complexa de paraplegia espástica hereditária caracterizada por início da infância até a idade adulta de espasticidade lentamente progressiva dos membros inferiores (resultando em distúrbios da marcha, respostas extensoras plantares e diminuição da sensação de vibração) associada a deficiência intelectual leve, ataxia cerebelar leve, neuropatia periférica (com amiotrofia distal dos membros superiores) e degeneração da retina. Corpo caloso fino é um achado de imagem comum.
Introdução
O que você precisa saber de cara
A paraplegia espástica autossômica recessiva tipo 15 é uma forma complexa de paraplegia espástica hereditária caracterizada por início da infância até a idade adulta de espasticidade lentamente progressiva dos membros inferiores (resultando em distúrbios da marcha, respostas extensoras plantares e diminuição da sensação de vibração) associada a deficiência intelectual leve, ataxia cerebelar leve, neuropatia periférica (com amiotrofia distal dos membros superiores) e degeneração da retina. Corpo caloso fino é um achado de imagem comum.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 15 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 56 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisPhosphatidylinositol 3-phosphate-binding protein required for the abscission step in cytokinesis: recruited to the midbody during cytokinesis and acts as a regulator of abscission. May also be required for efficient homologous recombination DNA double-strand break repair
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeMidbody
Spastic paraplegia 15, autosomal recessive
A form of spastic paraplegia, a neurodegenerative disorder characterized by a slow, gradual, progressive weakness and spasticity of the lower limbs. Rate of progression and the severity of symptoms are quite variable. Initial symptoms may include difficulty with balance, weakness and stiffness in the legs, muscle spasms, and dragging the toes when walking. In some forms of the disorder, bladder symptoms (such as incontinence) may appear, or the weakness and stiffness may spread to other parts of the body. SPG15 is a complex form associated with additional neurological symptoms such as cognitive deterioration or intellectual disability, axonal neuropathy, mild cerebellar signs, and, less frequently, a central hearing deficit, decreased visual acuity, or retinal degeneration.
Variantes genéticas (ClinVar)
619 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Paraplegia espástica autossômica recessiva tipo 15
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Retinal Findings in Kjellin Syndrome.
Keratoconus in hereditary spastic paraplegia 15 and Kjellin syndrome: a case report.
Hereditary spastic paraplegia 15 (HSP15) is a rare genetic disease manifesting with progressive muscle spasticity and paralysis of the lower limbs (paraplegia) caused by mutations in the ZFYVE26 gene. When spastic paraplegia is accompanied by retinal degeneration and cognitive impairment, it is known as Kjellin syndrome. We report on ocular manifestations in a case with HSP15 and Kjellin syndrome. We follow a 26-year-old male patient with HSP15 for 6 years. His condition was confirmed by clinical exome sequencing. In addition to regular systemic follow-ups, we performed ophthalmic examinations that included best corrected visual acuity, color vision testing, stereo acuity, visual fields, fundus photography, fundus autofluorescence, optical coherence tomography (OCT), OCT angiography, and corneal topography. Genetic testing in the patient revealed homozygosity for the pathogenic variant c.2114dupC; p. (Glu706Ter) in the ZFYVE26. Although the variant is present in population databases and ClinVar, this is the first report of this variant in HSP15 affected patient. During follow-up the patient's visual acuity declined. Ocular fundus findings comprised of slowly progressive retinal degeneration and a novel ocular phenotype in HSP15 - keratoconus. Corneal topography showed corneal thinning (thinnest location OD: 524 µm OS: 490 µm). Keratoconus was classified as RE: A1B2C0D1, LE: A3B4C1D3 according to Belin Keratoconus Staging System. We describe the clinical and ocular manifestations of a patient with HSP15 and Kjellin syndrome who was diagnosed with a pathogenic variant of ZFYVE26 gene. The patient developed keratoconus that to our knowledge is a novel ophthalmic phenotype in HSP15.
Late-onset Kjellin syndrome: Diagnosis of SPG11 on fundus examination.
Spastic paraplegia (SPG) is a heterogenous group of neurodegenerative disorders, that may include ocular involvement. Here we report the clinical data of a patient with late-onset Kjellin syndrome, a peculiar form of hereditary SPG with macular dystrophy. Clinical, functional and multimodal retinal imaging data were collected. Genetic testing was performed by Whole Exome Sequencing (WES). A 52-year-old female patient with SPG of unknown origin was referred for a progressive visual acuity loss. Multimodal fundus imaging revealed a peculiar macular dystrophy. Given the specific association of macular dystrophy and SPG, a Kjellin syndrome was suspected and genetic testing performed. WES revealed biallelic pathogenic variants in SPG11, co-segregating with disease in the family. Careful ophthalmological examination prompted the diagnosis and guided molecular testing. This case underlines the importance of a neuro-ophthalmologic assessment in patients with SPG.
Homocarnosinosis: A historical update and findings in the SPG11 gene.
A family with homocarnosinosis was reported in the literature in 1976. Three affected siblings had spastic paraplegia, retinitis pigmentosa, mental retardation, and cerebrospinal fluid (CSF) homocarnosine concentrations 20 times higher than in controls. Based on the clinical findings and new genetic techniques, we have been able to establish a precise genetic diagnosis. The medical records were re-evaluated, and genetic analyses were performed post-mortem in this original family. SNP array-based whole genome homozygosity mapping and Sanger sequencing of the SPG11 gene were performed. Seven additional Norwegian SPG11 patients and their disease-causing variants and clinical findings were evaluated. Homocarnosine levels in CSF were measured in four of these seven patients. A homozygous pathogenic splice-site variant in the SPG11 gene, c.2316 + 1G>A, was found. The clinical findings in the original family correlate with the heterogeneous SPG11 phenotype. The same variant was found in seven other Norwegian SPG11 patients, unrelated to the original family, either as homozygous or compound heterozygous constellation. Normal homocarnosine levels were found in the CSF of all unrelated SPG11 patients. A re-evaluation of the clinical symptoms and findings in the original family correlates with the SPG11 phenotype. The increased levels of homocarnosine do not seem to be a biomarker for SPG11 in our patients. Homocarnosinosis is still a biochemical aberration with unknown clinical significance.
Publicações recentes
Retinal Findings in Kjellin Syndrome.
Keratoconus in hereditary spastic paraplegia 15 and Kjellin syndrome: a case report.
Late-onset Kjellin syndrome: Diagnosis of SPG11 on fundus examination.
Homocarnosinosis: A historical update and findings in the SPG11 gene.
Kjellin syndrome: hereditary spastic paraplegia with pathognomonic macular appearance.
📚 EuropePMC10 artigos no totalmostrando 4
Retinal Findings in Kjellin Syndrome.
OphthalmologyKeratoconus in hereditary spastic paraplegia 15 and Kjellin syndrome: a case report.
Ophthalmic geneticsLate-onset Kjellin syndrome: Diagnosis of SPG11 on fundus examination.
European journal of ophthalmologyHomocarnosinosis: A historical update and findings in the SPG11 gene.
Acta neurologica ScandinavicaAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Paraplegia espástica autossômica recessiva tipo 15.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Paraplegia espástica autossômica recessiva tipo 15
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Retinal Findings in Kjellin Syndrome.
- Keratoconus in hereditary spastic paraplegia 15 and Kjellin syndrome: a case report.
- Late-onset Kjellin syndrome: Diagnosis of SPG11 on fundus examination.
- Homocarnosinosis: A historical update and findings in the SPG11 gene.
- Kjellin syndrome: hereditary spastic paraplegia with pathognomonic macular appearance.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:100996(Orphanet)
- OMIM OMIM:270700(OMIM)
- MONDO:0010044(MONDO)
- GARD:9581(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q32142628(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
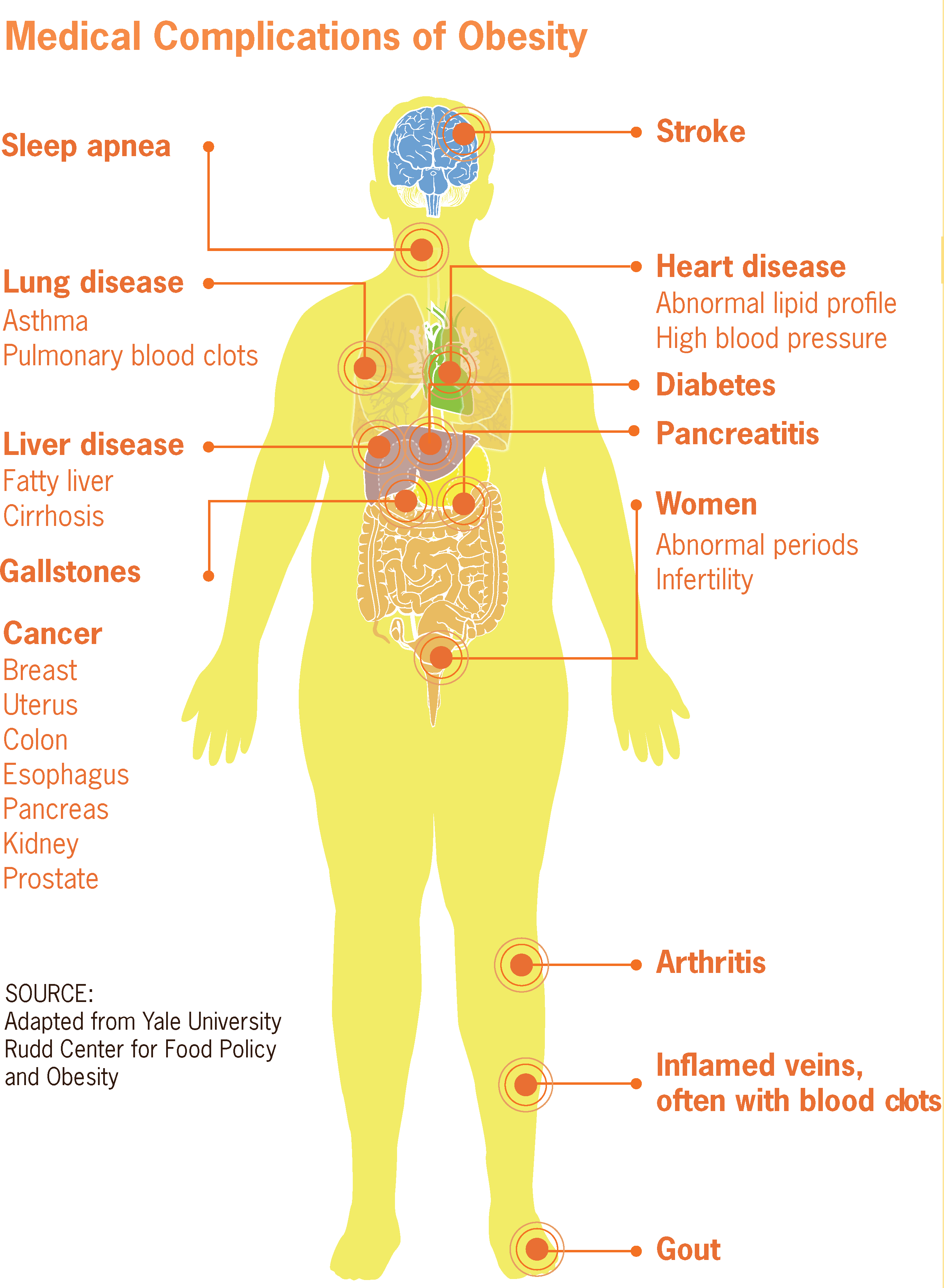
Paraplegia espástica autossômica recessiva tipo 15
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata