Uma microftalmia que faz parte de uma síndrome maior.
Introdução
O que você precisa saber de cara
Uma microftalmia que faz parte de uma síndrome maior.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 183 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 567 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
23 genes identificados com associação a esta condição.
Catalytic subunit of the Rab3 GTPase-activating (Rab3GAP) complex composed of RAB3GAP1 and RAB3GAP2, which accelerates the otherwise slow GTP hydrolysis catalyzed by Rab proteins (PubMed:9030515, PubMed:10859313, PubMed:39779760). Has GTPase-activating protein (GAP) activity towards various Rab3 subfamily members (RAB3A, RAB3B, RAB3C and RAB3D), RAB5A and RAB43 (PubMed:10859313, PubMed:9030515, PubMed:39779760). Additionally, it has guanine nucleotide exchange factor (GEF) activity towards RAB18
CytoplasmEndoplasmic reticulumGolgi apparatus, cis-Golgi network
Warburg micro syndrome 1
A rare, autosomal recessive syndrome characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe intellectual disability, spastic diplegia, and hypogonadism.
Ligand for members of the frizzled family of seven transmembrane receptors that functions in the canonical Wnt/beta-catenin signaling pathway (PubMed:30026314). Required for normal fusion of the chorion and the allantois during placenta development (By similarity). Required for central nervous system (CNS) angiogenesis and blood-brain barrier regulation (PubMed:30026314)
Secreted, extracellular space, extracellular matrixSecreted
Accessory subunit of the mitochondrial membrane respiratory chain NADH dehydrogenase (Complex I), that is believed not to be involved in catalysis. Complex I functions in the transfer of electrons from NADH to the respiratory chain. The immediate electron acceptor for the enzyme is believed to be ubiquinone
Mitochondrion inner membrane
Linear skin defects with multiple congenital anomalies 3
A disorder characterized by dermal, ocular, neurological and cardiac abnormalities. LSDMCA3 clinical features include linear skin defects on face and neck at birth, lacrimal duct atresia, myopia, nystagmus, strabismus, cardiomyopathy, axial hypotonia, seizures, corpus callosum agenesis, and dilation of lateral ventricles.
Multifunctional protein with various roles in different cellular compartments. May act in a redox sensitive manner. Associates with chromatin and binds DNA with a preference for non-canonical DNA structures such as single-stranded DNA. Can bend DNA and enhance DNA flexibility by looping thus providing a mechanism to promote activities on various gene promoters (By similarity). Proposed to be involved in the innate immune response to nucleic acids by acting as a cytoplasmic promiscuous immunogeni
NucleusChromosomeCytoplasm
Microphthalmia, syndromic, 13
A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS13 patients exhibit colobomatous microphthalmia with microcephaly, short stature, and psychomotor retardation.
ATP-dependent 5'-3' DNA helicase (PubMed:31253769, PubMed:8413672, PubMed:9771713). Component of the general transcription and DNA repair factor IIH (TFIIH) core complex, not absolutely essential for minimal transcription in vitro (PubMed:10024882, PubMed:17466626, PubMed:9771713). Required for transcription-coupled nucleotide excision repair (NER) of damaged DNA; recognizes damaged bases (PubMed:17466626, PubMed:23352696, PubMed:9771713). Sequestered in chromatin on UV-damaged DNA (PubMed:23352
NucleusCytoplasm, cytoskeleton, spindle
Xeroderma pigmentosum complementation group D
An autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. The skin develops marked freckling and other pigmentation abnormalities. Some XP-D patients present features of Cockayne syndrome, including cachectic dwarfism, pigmentary retinopathy, ataxia, decreased nerve conduction velocities. The phenotype combining xeroderma pigmentosum and Cockayne syndrome traits is referred to as XP-CS complex.
Transcription factor that may function in dorsoventral specification of the forebrain. Required for axon guidance and major tract formation in the developing forebrain. May contribute to the differentiation of the neuroretina, pigmented epithelium and optic stalk (By similarity)
Nucleus
Microphthalmia, syndromic, 11
A rare clinical entity including as main characteristics microphthalmia and small optic nerves, cleft lip and palate, absence of corpus callosum, hippocampal malformations, and absence of the pineal gland. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Essential factor involved in transcription-coupled nucleotide excision repair (TC-NER), a process during which RNA polymerase II-blocking lesions are rapidly removed from the transcribed strand of active genes (PubMed:16246722, PubMed:20541997, PubMed:22483866, PubMed:26620705, PubMed:32355176, PubMed:34526721, PubMed:38316879, PubMed:38600235, PubMed:38600236). Plays a central role in the initiation of the TC-NER process: specifically recognizes and binds RNA polymerase II stalled at a lesion,
NucleusChromosome
Cockayne syndrome B
A rare disorder characterized by cutaneous sensitivity to sunlight, abnormal and slow growth, cachectic dwarfism, progeroid appearance, progressive pigmentary retinopathy and sensorineural deafness. There is delayed neural development and severe progressive neurologic degeneration resulting in intellectual disability. Two clinical forms are recognized: in the classical form or Cockayne syndrome type 1, the symptoms are progressive and typically become apparent within the first few years or life; the less common Cockayne syndrome type 2 is characterized by more severe symptoms that manifest prenatally. Cockayne syndrome shows some overlap with certain forms of xeroderma pigmentosum. Unlike xeroderma pigmentosum, patients with Cockayne syndrome do not manifest increased freckling and other pigmentation abnormalities in the skin and have no significant increase in skin cancer.
Component of the cytochrome c oxidase, the last enzyme in the mitochondrial electron transport chain which drives oxidative phosphorylation. The respiratory chain contains 3 multisubunit complexes succinate dehydrogenase (complex II, CII), ubiquinol-cytochrome c oxidoreductase (cytochrome b-c1 complex, complex III, CIII) and cytochrome c oxidase (complex IV, CIV), that cooperate to transfer electrons derived from NADH and succinate to molecular oxygen, creating an electrochemical gradient over t
Mitochondrion inner membrane
Linear skin defects with multiple congenital anomalies 2
A distinct form of aplasia cutis congenita presenting as multiple linear skin defects on the face and neck associated with poor growth, microcephaly, and facial dysmorphism. Additional features include intellectual disability, nail dystrophy, short stature and cardiac abnormalities.
The small GTPases Rab are key regulators of intracellular membrane trafficking, from the formation of transport vesicles to their fusion with membranes (PubMed:24891604, PubMed:30970241). Rabs cycle between an inactive GDP-bound form and an active GTP-bound form that is able to recruit to membranes different sets of downstream effectors directly responsible for vesicle formation, movement, tethering and fusion (PubMed:24891604, PubMed:30970241). RAB18 is required for the localization of ZFYVE1 t
Endoplasmic reticulum membraneGolgi apparatus, cis-Golgi network membraneLipid dropletApical cell membrane
Warburg micro syndrome 3
A rare syndrome characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe intellectual disability, spastic diplegia, and hypogonadism.
Required for several aspects of embryonic development including normal development of the eye
NucleusCytoplasm
Microphthalmia/coloboma and skeletal dysplasia syndrome
A disease characterized by bilateral colobomatous microphthalmia or bilateral anophthalmia, associated with skeletal dysplasia in some cases. Additional ocular findings include microcornea, cataracts, corectopia and nystagmus. Intellectual disability is present in some patients.
Lyase that catalyzes the covalent linking of the heme group to the cytochrome C apoprotein to produce the mature functional cytochrome
Mitochondrion inner membraneMembrane
Linear skin defects with multiple congenital anomalies 1
A disorder characterized by dermal, ocular, neurological and cardiac abnormalities. LSDMCA1 main features are unilateral or bilateral microphthalmia, linear skin defects in affected females, and in utero lethality for males. Skin defects are limited to the face and neck, consisting of areas of aplastic skin that heal with age to form hyperpigmented areas. Additional features in female patients include agenesis of the corpus callosum, sclerocornea, chorioretinal abnormalities, infantile seizures, congenital heart defect, intellectual disability, and diaphragmatic hernia. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Non-catalytic component of a structure-specific DNA repair endonuclease responsible for the 5'-incision during DNA repair. Responsible, in conjunction with SLX4, for the first step in the repair of interstrand cross-links (ICL). Participates in the processing of anaphase bridge-generating DNA structures, which consist in incompletely processed DNA lesions arising during S or G2 phase, and can result in cytokinesis failure. Also required for homology-directed repair (HDR) of DNA double-strand bre
NucleusCytoplasm
Cerebro-oculo-facio-skeletal syndrome 4
A disorder of prenatal onset characterized by microcephaly, congenital cataracts, facial dysmorphism, neurogenic arthrogryposis, growth failure and severe psychomotor retardation. COFS is considered to be part of the nucleotide-excision repair disorders spectrum that include also xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome.
Receptor for retinoic acid. Retinoic acid receptors bind as heterodimers to their target response elements in response to their ligands, all-trans or 9-cis retinoic acid, and regulate gene expression in various biological processes. The RXR/RAR heterodimers bind to the retinoic acid response elements (RARE) composed of tandem 5'-AGGTCA-3' sites known as DR1-DR5. In the absence or presence of hormone ligand, acts mainly as an activator of gene expression due to weak binding to corepressors (PubMe
NucleusCytoplasm
Microphthalmia, syndromic, 12
A form of microphthalmia, a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities. MCOPS12 patients manifest variable features, including diaphragmatic hernia, pulmonary hypoplasia, and cardiac abnormalities.
Transcription factor probably involved in the development of the brain and the sense organs. Can bind to the bicoid/BCD target sequence (BTS): 5'-TCTAATCCC-3'
Nucleus
Microphthalmia, syndromic, 5
Patients manifest unilateral or bilateral microphthalmia/clinical anophthalmia and variable additional features including pituitary dysfunction, coloboma, microcornea, cataract, retinal dystrophy, hypoplasia or agenesis of the optic nerve, agenesis of the corpus callosum, developmental delay, joint laxity, hypotonia, and seizures. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Functions as a retinol transporter. Accepts all-trans retinol from the extracellular retinol-binding protein RBP4, facilitates retinol transport across the cell membrane, and then transfers retinol to the cytoplasmic retinol-binding protein RBP1 (PubMed:18316031, PubMed:22665496, PubMed:9452451). Retinol uptake is enhanced by LRAT, an enzyme that converts retinol to all-trans retinyl esters, the storage forms of vitamin A (PubMed:18316031, PubMed:22665496). Contributes to the activation of a sig
Cell membrane
Microphthalmia, syndromic, 9
A rare clinical entity including as main characteristics anophthalmia or severe microphthalmia, and pulmonary hypoplasia or aplasia. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Regulatory subunit of the Rab3 GTPase-activating (Rab3GAP) complex composed of RAB3GAP1 and RAB3GAP2, which accelerates the otherwise slow GTP hydrolysis catalyzed by Rab proteins (PubMed:9733780, PubMed:39779760). The complex has GTPase-activating protein (GAP) activity towards various Rab3 subfamily members (RAB3A, RAB3B, RAB3C and RAB3D), RAB5A and RAB43, and has guanine nucleotide exchange factor (GEF) activity towards RAB18 (PubMed:9733780, PubMed:39779760, PubMed:24891604). The Rab3GAP com
CytoplasmEndoplasmic reticulum
Martsolf syndrome 1
An autosomal recessive disease characterized by congenital cataracts, intellectual disability, and hypogonadism.
Growth factor of the TGF-beta superfamily that plays essential roles in many developmental processes, including neurogenesis, vascular development, angiogenesis and osteogenesis (PubMed:31363885). Acts in concert with PTHLH/PTHRP to stimulate ductal outgrowth during embryonic mammary development and to inhibit hair follicle induction (By similarity). Initiates the canonical BMP signaling cascade by associating with type I receptor BMPR1A and type II receptor BMPR2 (PubMed:25868050, PubMed:800600
Secreted, extracellular space, extracellular matrix
Microphthalmia, syndromic, 6
A disease characterized by microphthalmia/anophthalmia associated with facial, genital, skeletal, neurologic and endocrine anomalies. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Transcriptional corepressor. May specifically inhibit gene expression when recruited to promoter regions by sequence-specific DNA-binding proteins such as BCL6 and MLLT3. This repression may be mediated at least in part by histone deacetylase activities which can associate with this corepressor. Involved in the repression of TFAP2A; impairs binding of BCL6 and KDM2B to TFAP2A promoter regions. Via repression of TFAP2A acts as a negative regulator of osteo-dentiogenic capacity in adult stem cells
Nucleus
Microphthalmia, syndromic, 2
A very rare multiple congenital anomaly syndrome characterized by eye anomalies (congenital cataract, microphthalmia, or secondary glaucoma), facial abnormalities (long narrow face, high nasal bridge, pointed nose with cartilages separated at the tip, cleft palate, or submucous cleft palate), cardiac anomalies (atrial septal defect, ventricular septal defect, or floppy mitral valve) and dental abnormalities (canine radiculomegaly, delayed dentition, oligodontia, persistent primary teeth, or variable root length). Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
GTPase-activating protein (GAP) specific for Rab1 and Rab2 small GTPase families for which it can accelerate the intrinsic GTP hydrolysis rate by more than five orders of magnitude (PubMed:23236136). Also shows GAP activity for RAB18 GTPase (PubMed:26063829). Promotes RAB18 dissociation from the endoplasmic reticulum (ER) membrane into the cytosol, probably through stimulating RAB18 GTP-hydrolysis (PubMed:26063829). Involved in maintaining endoplasmic reticulum structure (PubMed:24891604)
Membrane
Warburg micro syndrome 4
A form of Warburg micro syndrome, a rare syndrome characterized by microcephaly, microphthalmia, microcornia, congenital cataracts, optic atrophy, cortical dysplasia, in particular corpus callosum hypoplasia, severe intellectual disability, spastic diplegia, and hypogonadism.
DNA-binding protein that binds to the 5'-CAAG-3' core sequence. May function as a transcriptional repressor. Seems to act as a transcriptional antagonist of NKX2-5. May play an important role in the development of craniofacial structures such as the eye and ear
Nucleus
Oculoauricular syndrome
A syndrome characterized by microphthalmia, microcornea, anterior segment dysgenesis, cataract, ocular coloboma, retinal pigment epithelium abnormalities, rod-cone dystrophy, and anomalies of the external ear.
Transcription factor that forms a trimeric complex with OCT4 on DNA and controls the expression of a number of genes involved in embryonic development such as YES1, FGF4, UTF1 and ZFP206 (By similarity). Binds to the proximal enhancer region of NANOG (By similarity). Critical for early embryogenesis and for embryonic stem cell pluripotency (PubMed:18035408). Downstream SRRT target that mediates the promotion of neural stem cell self-renewal (By similarity). Keeps neural cells undifferentiated by
Nucleus speckleCytoplasmNucleus
Microphthalmia, syndromic, 3
A disease characterized by the rare association of malformations including uni- or bilateral anophthalmia or microphthalmia, and esophageal atresia with trachoesophageal fistula. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Single-stranded structure-specific DNA endonuclease involved in DNA excision repair (PubMed:32522879, PubMed:32821917, PubMed:7651464, PubMed:8078765, PubMed:8090225, PubMed:8206890). Makes the 3'incision in DNA nucleotide excision repair (NER) (PubMed:32522879, PubMed:32821917, PubMed:8078765, PubMed:8090225). Binds and bends DNA repair bubble substrate and breaks base stacking at the single-strand/double-strand DNA junction of the DNA bubble (PubMed:32522879). Plays a role in base excision rep
NucleusChromosome
Xeroderma pigmentosum complementation group G
An autosomal recessive pigmentary skin disorder characterized by solar hypersensitivity of the skin, high predisposition for developing cancers on areas exposed to sunlight and, in some cases, neurological abnormalities. The skin develops marked freckling and other pigmentation abnormalities. Some XP-G patients present features of Cockayne syndrome, cachectic dwarfism, pigmentary retinopathy, ataxia, decreased nerve conduction velocities. The phenotype combining xeroderma pigmentosum and Cockayne syndrome traits is referred to as XP-CS complex.
Catalytic subunit of N-terminal acetyltransferase complexes which display alpha (N-terminal) acetyltransferase activity (PubMed:15496142, PubMed:19420222, PubMed:19826488, PubMed:20145209, PubMed:20154145, PubMed:25489052, PubMed:27708256, PubMed:29754825, PubMed:32042062). Acetylates amino termini that are devoid of initiator methionine (PubMed:19420222). The alpha (N-terminal) acetyltransferase activity may be important for vascular, hematopoietic and neuronal growth and development. Without N
CytoplasmNucleus
N-terminal acetyltransferase deficiency
An enzymatic deficiency resulting in postnatal growth failure with severe delays and dysmorphic features. It is clinically characterized by wrinkled forehead, prominent eyes, widely opened anterior and posterior fontanels, downsloping palpebral fissures, thickened lids, large ears, flared nares, hypoplastic alae, short columella, protruding upper lip, and microretrognathia. There are also delayed closing of fontanels and broad great toes. Skin is characterized by redundancy or laxity with minimal subcutaneous fat, cutaneous capillary malformations, and very fine hair and eyebrows. Death results from cardiogenic shock following arrhythmia.
Variantes genéticas (ClinVar)
380 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
74 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de microftalmia sindromática-anoftalmia-coloboma
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Molecular investigation in individuals with orofacial clefts and microphthalmia-anophthalmia-coloboma spectrum.
Este estudo investigou a causa genética em indivíduos com fendas orofaciais e malformações oculares graves (microftalmia-anoftalmia-coloboma), mostrando que o Sequenciamento do Exoma Completo (WES) foi a abordagem mais eficaz. O WES permitiu um diagnóstico conclusivo em mais de um terço dos pacientes, identificando variantes em genes como CHD7, TFAP2A, POMT1, PTPN11 e TP63, associadas a síndromes conhecidas (ex: CHARGE, LEOPARD). Isso reforça a complexidade genética dessas condições e a importância de investigar genes relacionados a doenças dos cílios para um diagnóstico mais preciso e aconselhamento genético adequado.
🇧🇷 traduzidoCommercial Gene Panels for Congenital Anterior Segment Anomalies: Are They All the Same?
Este estudo comparou painéis genéticos comerciais para anomalias oculares congênitas como a microftalmia-anoftalmia-coloboma (MAC), revelando uma grande e significativa variabilidade na composição dos genes testados entre os laboratórios. Para médicos e pacientes, isso implica que a escolha do painel e a interpretação dos resultados são complexas, pois muitos genes incluídos são menos estudados, gerando incerteza sobre o diagnóstico e a causa da condição. Isso enfatiza a necessidade de estudos mais rigorosos para padronizar e orientar a seleção de painéis genéticos eficazes.
🇧🇷 traduzidoSpectrum of congenital and inherited ocular disorders seen in a genetic clinic: Experience of a developing ocular genetic service.
Este estudo destaca que o espectro de microftalmia-anoftalmia-coloboma (MAC) é uma condição ocular comum em clínicas genéticas, sendo frequentemente parte de síndromes com outras manifestações. Para pacientes e médicos, é essencial considerar a natureza sindrômica do MAC, buscando outras condições associadas. A investigação genética é uma ferramenta valiosa, pois pode identificar a causa e oferecer a oportunidade de diagnóstico pré-natal, auxiliando no planejamento familiar.
🇧🇷 traduzidoA 300-kb microduplication of 7q36.3 in a patient with triphalangeal thumb-polysyndactyly syndrome combined with congenital heart disease and optic disc coloboma: a case report.
Este estudo de caso descreve uma apresentação rara da síndrome de polegar tri-falângico com polissindactilia (TPT-PS), que geralmente afeta apenas os membros, mas que se manifestou aqui com doença cardíaca congênita grave e coloboma ocular. A análise genética revelou uma microduplicação de 7q36.3, conhecida por causar TPT-PS, neste paciente complexo. Isso sugere que esta alteração genética pode ter um impacto fenotípico mais amplo do que o previamente reconhecido, incluindo malformações cardíacas e oculares, sendo um alerta importante para médicos e pacientes sobre a possível associação do 7q36.3 com manifestações sistêmicas além das malformações de membros.
🇧🇷 traduzidoIdentification of a possible association of JAK2 in development of microphthalmia, anophthalmia, and coloboma (MAC) complex in a child with 9p deletion and duplication.
Este artigo relata o primeiro caso de Microftalmia, Anoſtalmia e Coloboma (MAC) – um espectro de anomalias oculares – associado a uma deleção e duplicação no cromossomo 9p, em uma criança que também apresentava outras condições sistêmicas (como problemas cardíacos e renais). Para pacientes com MAC, especialmente aqueles com outras malformações, isso reforça a importância da investigação genética detalhada, agora incluindo a região 9p. A duplicação do gene JAK2, localizado nessa região, é proposta como um possível fator que pode ter contribuído para o desenvolvimento do MAC.
🇧🇷 traduzidoPublicações recentes
Familial co-occurrence of autism spectrum disorder and 47 XYY syndrome: revisiting the role of Y chromosome dosage in neurodevelopment.
Prenatal detection of Gorlin-Goltz syndrome: a case report and focused review of the literature.
Epidemiological characteristics and co-occurrence patterns of Rothia species and respiratory pathogens: from population surveillance to mechanistic insights.
Sirolimus for Extracranial Arteriovenous Malformations: A Scoping Review of the Evidence in Syndromic and Non-Syndromic Cases.
Dual disorders: an overview.
📚 EuropePMCmostrando 7
Molecular investigation in individuals with orofacial clefts and microphthalmia-anophthalmia-coloboma spectrum.
European journal of human genetics : EJHGCommercial Gene Panels for Congenital Anterior Segment Anomalies: Are They All the Same?
American journal of ophthalmologySpectrum of congenital and inherited ocular disorders seen in a genetic clinic: Experience of a developing ocular genetic service.
Indian journal of ophthalmologyA 300-kb microduplication of 7q36.3 in a patient with triphalangeal thumb-polysyndactyly syndrome combined with congenital heart disease and optic disc coloboma: a case report.
BMC medical genomicsIdentification of a possible association of JAK2 in development of microphthalmia, anophthalmia, and coloboma (MAC) complex in a child with 9p deletion and duplication.
Ophthalmic genetics[Clinical and Genetic Characteristics of Ocular Developmental Disorders: MAC-Spectrum, Anterior Segment Dysgenesis].
Klinische Monatsblatter fur AugenheilkundeThe Oculome Panel Test: Next-Generation Sequencing to Diagnose a Diverse Range of Genetic Developmental Eye Disorders.
OphthalmologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de microftalmia sindromática-anoftalmia-coloboma.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de microftalmia sindromática-anoftalmia-coloboma
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Molecular investigation in individuals with orofacial clefts and microphthalmia-anophthalmia-coloboma spectrum.
- Commercial Gene Panels for Congenital Anterior Segment Anomalies: Are They All the Same?
- Spectrum of congenital and inherited ocular disorders seen in a genetic clinic: Experience of a developing ocular genetic service.
- A 300-kb microduplication of 7q36.3 in a patient with triphalangeal thumb-polysyndactyly syndrome combined with congenital heart disease and optic disc coloboma: a case report.
- Identification of a possible association of JAK2 in development of microphthalmia, anophthalmia, and coloboma (MAC) complex in a child with 9p deletion and duplication.
- Familial co-occurrence of autism spectrum disorder and 47 XYY syndrome: revisiting the role of Y chromosome dosage in neurodevelopment.
- Prenatal detection of Gorlin-Goltz syndrome: a case report and focused review of the literature.
- Epidemiological characteristics and co-occurrence patterns of Rothia species and respiratory pathogens: from population surveillance to mechanistic insights.
- Sirolimus for Extracranial Arteriovenous Malformations: A Scoping Review of the Evidence in Syndromic and Non-Syndromic Cases.
- Dual disorders: an overview.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:202948(Orphanet)
- MONDO:0016073(MONDO)
- GARD:20342(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q29982037(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
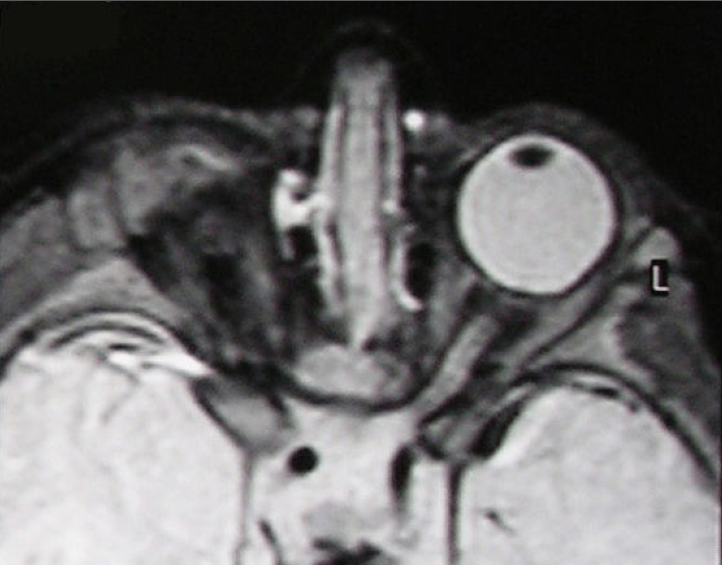
Síndrome de microftalmia sindromática-anoftalmia-coloboma
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata