Doença de Moyamoya - baixa estatura - traços faciais atípicos - hipogonadismo (problema hormonal) com estímulo cerebral elevado é uma síndrome muito rara, hereditária, neurológica e dismórfica (que causa alterações na aparência física), caracterizada pela doença de Moyamoya, baixa estatura que começa a ser notada após o nascimento, e traços faciais atípicos e bem definidos.
Introdução
O que você precisa saber de cara
Doença de Moyamoya - baixa estatura - traços faciais atípicos - hipogonadismo (problema hormonal) com estímulo cerebral elevado é uma síndrome muito rara, hereditária, neurológica e dismórfica (que causa alterações na aparência física), caracterizada pela doença de Moyamoya, baixa estatura que começa a ser notada após o nascimento, e traços faciais atípicos e bem definidos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 44 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: X-linked recessive.
Metalloprotease that specifically cleaves 'Lys-63'-linked polyubiquitin chains (PubMed:19214193, PubMed:20656690, PubMed:24075985, PubMed:26344097). Does not have activity toward 'Lys-48'-linked polyubiquitin chains (PubMed:19214193, PubMed:20656690, PubMed:24075985, PubMed:26344097). Component of the BRCA1-A complex, a complex that specifically recognizes 'Lys-63'-linked ubiquitinated histones H2A and H2AX at DNA lesions sites, leading to target the BRCA1-BARD1 heterodimer to sites of DNA damag
NucleusCytoplasmCytoplasm, cytoskeleton, spindle pole
Variantes genéticas (ClinVar)
240 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de angiopatia de Moyamoya-baixa estatura-dismorfia facial-hipogonadismo hipergonadotrópico
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
BRCC3 -Associated Syndromic Moyamoya Angiopathy Diagnosed Through Clinical RNA Sequencing.
Moyamoya angiopathy is a cerebral vasculopathy causing progressive stenosis of the internal carotid arteries and the compensatory development of collateral blood vessels, leading to brain ischemia and an increased risk of cerebral haemorrhage. Although multiple non-genetic causes have been associated with moyamoya syndrome, it can also be associated with rare genetic syndromes. Moyamoya Disease 4, characterised by a short stature, hypergonadotropic hypogonadism and facial dysmorphism (MYMY4, OMIM #300845), also referred to as BRCC3-associated moyamoya syndrome, has so far been described in 11 individuals. Here, we describe a 23-year-old male presenting with moyamoya syndrome, global developmental delay and intellectual disability, epilepsy, short stature and dysmorphic features, who after > 17 years of uninformative diagnostics was diagnosed with BRCC3-associated moyamoya syndrome after clinical RNA-seq. Transcriptome analysis showed reduced expression of the likely disease-causing gene BRCC3 in patient-derived fibroblasts, which was subsequently found to be caused by a ~ 26 kb Xq28 deletion. We furthermore review all reported cases of BRCC3-associated moyamoya syndrome, further delineating this clinical entity.
Severe Hemophilia A and Moyamoya Syndrome in a 19-Year-Old Boy Caused by Xq28 Microdeletion.
Severe hemophilia A and moyamoya (SHAM) syndrome is a rare condition that combines hemophilia A and moyamoya disease (MMD) due to an Xq28 microdeletion encompassing the F8 and BRCC3 genes. Here, we report the case of a 19-year-old male patient with hemophilia A and hypogonadism that presented with right-sided hemiparesis and dysarthria. Brain magnetic resonance imaging and angiography revealed an ischemic lesion in the left lobe and stenosis of both middle cerebral arteries with a concomitant thick vascular network, compatible with moyamoya disease. Next-generation sequence revealed a large Xq28 deletion compatible with SHAM syndrome. The patient was treated with acetylsalicylic acid and neurosurgical intervention was scheduled. Our patient is one of the few cases reported in the literature with Xq28 microdeletion encompassing the F8, hemophilia A causative gene, and BRCC3, responsible for MMD, presenting with a compound phenotype that included neurological manifestations and hypogonadism. In conclusion, diagnosis of MMD should be considered in any male, young patient with symptoms of ischemic stroke with no obvious explanation, and especially in patients with known hemophilia, since a relationship between the two conditions has been documented.
Moyamoya angiopathy in a case of Klinefelter syndrome.
Moyamoya angiopathy, a rare cerebrovascular condition, can be primary (moyamoya disease) or secondary (moyamoya syndrome). Genetic factors, such as the ring finger protein 213 (RNF213), have been associated with moyamoya disease. However, X-linked moyamoya angiopathy/moyamoya syndrome and hypergonadotropic hypogonadism associated with moyamoya syndrome are rare. We report a case of a 14-year-old boy who presented with transient bilateral hemiparesis, recurrent seizures and cognitive decline. He previously had surgery for left-sided cryptorchidism and had been diagnosed with "epileptic attacks" or "functional movement disorders" in previous hospital admissions. Magnetic resonance angiography of the brain showed narrowing of supraclinoid portion of internal carotid arteries, as well as of middle and anterior cerebral arteries, and the presence of multiple collaterals. These findings were suggestive of moyamoya angiopathy. Laboratory investigations and karyotyping revealed a diagnosis of Klinefelter syndrome. This case presents a unique association of moyamoya angiopathy and Klinefelter syndrome in a boy from a poor socio-economic background, where the diagnosis and adequate treatment were delayed due to a lack of awareness and expertise.
Publicações recentes
Moyamoya angiopathy in a case of Klinefelter syndrome.
Empty sella, hypogonadism and hypopituitarism secondary to moyamoya disease.
📚 EuropePMCmostrando 3
BRCC3 -Associated Syndromic Moyamoya Angiopathy Diagnosed Through Clinical RNA Sequencing.
Clinical geneticsSevere Hemophilia A and Moyamoya Syndrome in a 19-Year-Old Boy Caused by Xq28 Microdeletion.
Case reports in neurologyMoyamoya angiopathy in a case of Klinefelter syndrome.
Child's nervous system : ChNS : official journal of the International Society for Pediatric NeurosurgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de angiopatia de Moyamoya-baixa estatura-dismorfia facial-hipogonadismo hipergonadotrópico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de angiopatia de Moyamoya-baixa estatura-dismorfia facial-hipogonadismo hipergonadotrópico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- BRCC3 -Associated Syndromic Moyamoya Angiopathy Diagnosed Through Clinical RNA Sequencing.
- Severe Hemophilia A and Moyamoya Syndrome in a 19-Year-Old Boy Caused by Xq28 Microdeletion.
- Moyamoya angiopathy in a case of Klinefelter syndrome.Child's nervous system : ChNS : official journal of the International Society for Pediatric Neurosurgery· 2022· PMID 34628529mais citado
- Empty sella, hypogonadism and hypopituitarism secondary to moyamoya disease.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:280679(Orphanet)
- OMIM OMIM:300845(OMIM)
- MONDO:0010448(MONDO)
- GARD:17301(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1951267(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
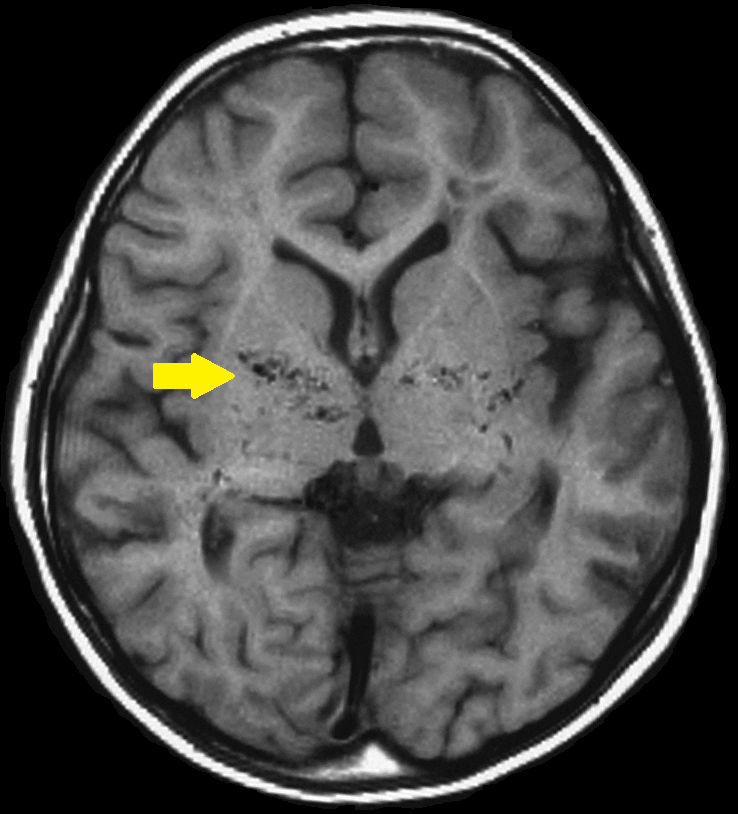
Síndrome de angiopatia de Moyamoya-baixa estatura-dismorfia facial-hipogonadismo hipergonadotrópico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata