A ataxia cerebelar autossômica recessiva associada à espectrina é uma doença neurológica genética rara, causada por mutações no gene SPTBN2. Ela se caracteriza por um atraso global no desenvolvimento já na primeira infância (fase de bebê), seguido, na infância (período de criança), por dificuldade de coordenação ao andar (ataxia da marcha), problemas em controlar a precisão dos movimentos dos braços e pernas (dismetria) e dificuldade para realizar movimentos rápidos e repetitivos (disdiadococinesia). Inclui também deficiência intelectual de leve a grave, atrofia do cerebelo (o cerebelo, parte do cérebro responsável pela coordenação, se deteriora), e movimentos oculares anormais, como: desvio de um olho para dentro (estrabismo convergente), movimentos rápidos dos olhos que não alcançam o alvo (sacadas hipométricas), dificuldade em seguir objetos em movimento de forma suave (movimentos de perseguição irregulares) e amplitude de movimento dos olhos limitada.
Introdução
O que você precisa saber de cara
A ataxia cerebelar autossômica recessiva associada à espectrina é uma doença neurológica genética rara, causada por mutações no gene SPTBN2. Ela se caracteriza por um atraso global no desenvolvimento já na primeira infância (fase de bebê), seguido, na infância (período de criança), por dificuldade de coordenação ao andar (ataxia da marcha), problemas em controlar a precisão dos movimentos dos braços e pernas (dismetria) e dificuldade para realizar movimentos rápidos e repetitivos (disdiadococinesia). Inclui também deficiência intelectual de leve a grave, atrofia do cerebelo (o cerebelo, parte do cérebro responsável pela coordenação, se deteriora), e movimentos oculares anormais, como: desvio de um olho para dentro (estrabismo convergente), movimentos rápidos dos olhos que não alcançam o alvo (sacadas hipométricas), dificuldade em seguir objetos em movimento de forma suave (movimentos de perseguição irregulares) e amplitude de movimento dos olhos limitada.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
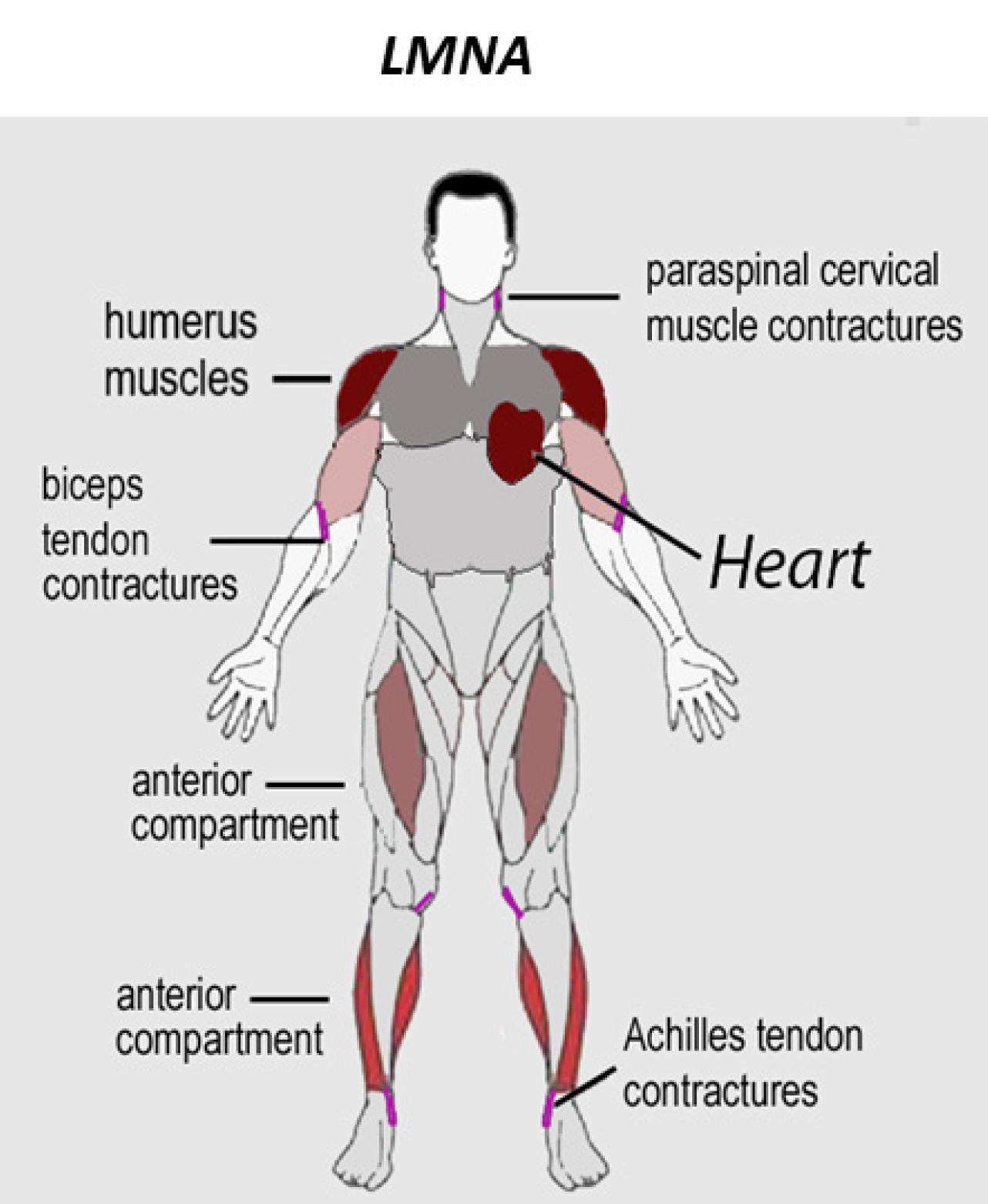
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 27 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisProbably plays an important role in neuronal membrane skeleton
Cytoplasm, cytoskeletonCytoplasm, cell cortex
Spinocerebellar ataxia 5
Spinocerebellar ataxia is a clinically and genetically heterogeneous group of cerebellar disorders. Patients show progressive incoordination of gait and often poor coordination of hands, speech and eye movements, due to degeneration of the cerebellum with variable involvement of the brainstem and spinal cord. SCA5 is an autosomal dominant cerebellar ataxia (ADCA). It is a slowly progressive disorder with variable age at onset, ranging between 10 and 50 years.
Variantes genéticas (ClinVar)
260 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Ataxia cerebelosa autossômica recessiva associada à espectrina
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration.
(1) Background: A non-progressive congenital ataxia (NPCA) phenotype caused by β-III spectrin (SPTBN2) mutations has emerged, mimicking spinocerebellar ataxia, autosomal recessive type 14 (SCAR14). The pattern of inheritance, however, resembles that of autosomal dominant classical spinocerebellar ataxia type 5 (SCA5). (2) Methods: In-depth phenotyping of two boys studied by a customized gene panel. Candidate variants were sought by structural modeling and protein expression. An extensive review of the literature was conducted in order to better characterize the SPTBN2-associated NPCA. (3) Results: Patients exhibited an NPCA with hypotonia, developmental delay, cerebellar syndrome, and cognitive deficits. Both probands presented with progressive global cerebellar volume loss in consecutive cerebral magnetic resonance imaging studies, characterized by decreasing midsagittal vermis relative diameter measurements. Cortical hyperintensities were observed on fluid-attenuated inversion recovery (FLAIR) images, suggesting a neurodegenerative process. Each patient carried a novel de novo SPTBN2 substitution: c.193A > G (p.K65E) or c.764A > G (p.D255G). Modeling and protein expression revealed that both mutations might be deleterious. (4) Conclusions: The reported findings contribute to a better understanding of the SPTBN2-associated phenotype. The mutations may preclude proper structural organization of the actin spectrin-based membrane skeleton, which, in turn, is responsible for the underlying disease mechanism.
βIII Spectrin Is Necessary for Formation of the Constricted Neck of Dendritic Spines and Regulation of Synaptic Activity in Neurons.
Dendritic spines are postsynaptic structures in neurons often having a mushroom-like shape. Physiological significance and cytoskeletal mechanisms that maintain this shape are poorly understood. The spectrin-based membrane skeleton maintains the biconcave shape of erythrocytes, but whether spectrins also determine the shape of nonerythroid cells is less clear. We show that βIII spectrin in hippocampal and cortical neurons from rodent embryos of both sexes is distributed throughout the somatodendritic compartment but is particularly enriched in the neck and base of dendritic spines and largely absent from spine heads. Electron microscopy revealed that βIII spectrin forms a detergent-resistant cytoskeletal network at these sites. Knockdown of βIII spectrin results in a significant decrease in the density of dendritic spines. Surprisingly, the density of presynaptic terminals is not affected by βIII spectrin knockdown. However, instead of making normal spiny synapses, the presynaptic structures in βIII spectrin-depleted neurons make shaft synapses that exhibit increased amplitudes of miniature EPSCs indicative of excessive postsynaptic excitation. Thus, βIII spectrin is necessary for formation of the constricted shape of the spine neck, which in turn controls communication between the synapse and the parent dendrite to prevent excessive excitation. Notably, mutations of SPTNB2 encoding βIII spectrin are associated with neurodegenerative syndromes, spinocerebellar ataxia Type 5, and spectrin-associated autosomal recessive cerebellar ataxia Type 1, but molecular mechanisms linking βIII spectrin functions to neuronal pathologies remain unresolved. Our data suggest that spinocerebellar ataxia Type 5 and spectrin-associated autosomal recessive cerebellar ataxia Type 1 pathology likely arises from poorly controlled synaptic activity that leads to excitotoxicity and neurodegeneration.SIGNIFICANCE STATEMENT Dendritic spines are small protrusions from neuronal dendrites that make synapses with axons of other neurons in the brain. Dendritic spines usually have a mushroom-like shape, which is essential for brain functions, because aberrant spine morphology is associated with many neuropsychiatric disorders. The bulbous head of a mushroom-shaped spine makes the synapse, whereas the narrow neck transmits the incoming signals to the dendrite and supposedly controls the signal propagation. We show that a cytoskeletal protein βIII spectrin plays a key role for the formation of narrow spine necks. In the absence of βIII spectrin, dendritic spines collapse onto dendrites. As a result, synaptic strength exceeds acceptable levels and damages neurons, explaining pathology of human syndromes caused by βIII spectrin mutations.
Posterior cerebellar Purkinje cells in an SCA5/SPARCA1 mouse model are especially vulnerable to the synergistic effect of loss of β-III spectrin and GLAST.
Clinical phenotypes of spinocerebellar ataxia type-5 (SCA5) and spectrin-associated autosomal recessive cerebellar ataxia type-1 (SPARCA1) are mirrored in mice lacking β-III spectrin (β-III-/-). One function of β-III spectrin is the stabilization of the Purkinje cell-specific glutamate transporter EAAT4 at the plasma membrane. In β-III-/- mice EAAT4 levels are reduced from an early age. In contrast levels of the predominant cerebellar glutamate transporter GLAST, expressed in Bergmann glia, only fall progressively from 3 months onwards. Here we elucidated the roles of these two glutamate transporters in cerebellar pathogenesis mediated through loss of β-III spectrin function by studying EAAT4 and GLAST knockout mice as well as crosses of both with β-III-/- mice. Our data demonstrate that EAAT4 loss, but not abnormal AMPA receptor composition, in young β-III-/- mice underlies early Purkinje cell hyper-excitability and that subsequent loss of GLAST, superimposed on the earlier deficiency of EAAT4, is responsible for Purkinje cell loss and progression of motor deficits. Yet the loss of GLAST appears to be independent of EAAT4 loss, highlighting that other aspects of Purkinje cell dysfunction underpin the pathogenic loss of GLAST. Finally, our results demonstrate that Purkinje cells in the posterior cerebellum of β-III-/- mice are most susceptible to the combined loss of EAAT4 and GLAST, with degeneration of proximal dendrites, the site of climbing fibre innervation, most pronounced. This highlights the necessity for efficient glutamate clearance from these regions and identifies dysregulation of glutamatergic neurotransmission particularly within the posterior cerebellum as a key mechanism in SCA5 and SPARCA1 pathogenesis.
Cerebellar ataxias: β-III spectrin's interactions suggest common pathogenic pathways.
Spinocerebellar ataxias (SCAs) are a genetically heterogeneous group of disorders all characterised by postural abnormalities, motor deficits and cerebellar degeneration. Animal and in vitro models have revealed β-III spectrin, a cytoskeletal protein present throughout the soma and dendritic tree of cerebellar Purkinje cells, to be required for the maintenance of dendritic architecture and for the trafficking and/or stabilisation of several membrane proteins: ankyrin-R, cell adhesion molecules, metabotropic glutamate receptor-1 (mGluR1), voltage-gated sodium channels (Nav ) and glutamate transporters. This scaffold of interactions connects β-III spectrin to a wide variety of proteins implicated in the pathology of many SCAs. Heterozygous mutations in the gene encoding β-III spectrin (SPTBN2) underlie SCA type-5 whereas homozygous mutations cause spectrin associated autosomal recessive ataxia type-1 (SPARCA1), an infantile form of ataxia with cognitive impairment. Loss-of β-III spectrin function appears to underpin cerebellar dysfunction and degeneration in both diseases resulting in thinner dendrites, excessive dendritic protrusion with loss of planarity, reduced resurgent sodium currents and abnormal glutamatergic neurotransmission. The initial physiological consequences are a decrease in spontaneous activity and excessive excitation, likely to be offsetting each other, but eventually hyperexcitability gives rise to dark cell degeneration and reduced cerebellar output. Similar molecular mechanisms have been implicated for SCA1, 2, 3, 7, 13, 14, 19, 22, 27 and 28, highlighting alterations to intrinsic Purkinje cell activity, dendritic architecture and glutamatergic transmission as possible common mechanisms downstream of various loss-of-function primary genetic defects. A key question for future research is whether similar mechanisms underlie progressive cerebellar decline in normal ageing.
Publicações recentes
Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration.
Posterior cerebellar Purkinje cells in an SCA5/SPARCA1 mouse model are especially vulnerable to the synergistic effect of loss of β-III spectrin and GLAST.
Cerebellar ataxias: β-III spectrin's interactions suggest common pathogenic pathways.
β-III spectrin underpins ankyrin R function in Purkinje cell dendritic trees: protein complex critical for sodium channel activity is impaired by SCA5-associated mutations.
Recessive mutations in SPTBN2 implicate β-III spectrin in both cognitive and motor development.
📚 EuropePMCmostrando 4
Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration.
International journal of molecular sciencesβIII Spectrin Is Necessary for Formation of the Constricted Neck of Dendritic Spines and Regulation of Synaptic Activity in Neurons.
The Journal of neuroscience : the official journal of the Society for NeurosciencePosterior cerebellar Purkinje cells in an SCA5/SPARCA1 mouse model are especially vulnerable to the synergistic effect of loss of β-III spectrin and GLAST.
Human molecular geneticsCerebellar ataxias: β-III spectrin's interactions suggest common pathogenic pathways.
The Journal of physiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Ataxia cerebelosa autossômica recessiva associada à espectrina.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Ataxia cerebelosa autossômica recessiva associada à espectrina
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Expanding the β-III Spectrin-Associated Phenotypes toward Non-Progressive Congenital Ataxias with Neurodegeneration.
- βIII Spectrin Is Necessary for Formation of the Constricted Neck of Dendritic Spines and Regulation of Synaptic Activity in Neurons.The Journal of neuroscience : the official journal of the Society for Neuroscience· 2017· PMID 28576936mais citado
- Posterior cerebellar Purkinje cells in an SCA5/SPARCA1 mouse model are especially vulnerable to the synergistic effect of loss of β-III spectrin and GLAST.
- Cerebellar ataxias: β-III spectrin's interactions suggest common pathogenic pathways.
- β-III spectrin underpins ankyrin R function in Purkinje cell dendritic trees: protein complex critical for sodium channel activity is impaired by SCA5-associated mutations.
- Recessive mutations in SPTBN2 implicate β-III spectrin in both cognitive and motor development.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:352403(Orphanet)
- OMIM OMIM:615386(OMIM)
- MONDO:0014159(MONDO)
- GARD:17516(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21124567(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Ataxia cerebelosa autossômica recessiva associada à espectrina
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata