A síndrome de Joubert com distrofia torácica asfixiante de Jeune (JATD) é uma doença óssea genética extremamente rara caracterizada pelas características clássicas da síndrome de Joubert (ou seja, malformação do tronco cerebral causando ataxia, hipotonia, comprometimento cognitivo e movimentos oculares anormais), associada às anomalias esqueléticas encontradas na JATD, incluindo displasia de costelas curtas e tórax estreito, causando insuficiência respiratória, membros curtos e alterações metafisárias.
Introdução
O que você precisa saber de cara
A síndrome de Joubert com distrofia torácica asfixiante de Jeune (JATD) é uma doença óssea genética extremamente rara caracterizada pelas características clássicas da síndrome de Joubert (ou seja, malformação do tronco cerebral causando ataxia, hipotonia, comprometimento cognitivo e movimentos oculares anormais), associada às anomalias esqueléticas encontradas na JATD, incluindo displasia de costelas curtas e tórax estreito, causando insuficiência respiratória, membros curtos e alterações metafisárias.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 44 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 118 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
May play a role in cell-cycle-dependent microtubule organization
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, spindle poleCell projection, cilium
Joubert syndrome 21
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Required for ciliogenesis and sonic hedgehog/SHH signaling. Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1. May play a role in early ciliogenesis in the disappearance of centriolar satellites that preceeds ciliary vesicle formation (PubMed:24421332). Involved in regulation of cell intracellular organization. Involved in regulation of cell polarity (By similarity). Required for asymmetrical localization of CEP
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomePhotoreceptor inner segmentCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 23
A mild form of Joubert syndrome, a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Variantes genéticas (ClinVar)
516 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 4 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de Joubert com distrofia torácica asfixiante de Jeune
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Fetal ciliopathies: a retrospective observational single-center study.
Report on the diagnosis of prenatally suspected multisystem ciliopathies in a single center between 2002 and 2020. Retrospective observational single-center study including pregnancies with prenatal ultrasound features of multisystem ciliopathies, such as hyperechogenic kidneys together with polydactyly and/or other skeletal and extraskeletal findings. Cases were compared according to their prenatal findings and outcomes. 36 cases of multisystem ciliopathies were diagnosed. Meckel-Gruber syndrome (MKS) was the most common ciliopathy (n = 19/36, 52.8%), followed by disorders that belong to the group of short-rib thoracic dysplasia (SRTD, n = 10/36, 27.8%) McKusick-Kaufmann syndrome (MKKS, n = 4/36, 11.1%), Bardet-Biedl syndrome (BBS, n = 2/36, 5.5%) and Joubert syndrome (n = 1/36, 2.8%). All cases showed abnormalities of the kidneys, most often hyperechogenic parenchyma (n = 26/36, 72.2%), cystic dysplasia (n = 24/36, 66.7%), and/or bilateral kidney enlargement (n = 22/36, 61.1%). Oligohydramnios was mainly present in fetuses with MKS. Polydactyly (n = 18/36), abnormalities of the CNS (n = 25/36), and heart defects (n = 10/36) were associated in 50%, 69.4%, and 27.8%, respectively. Prenatal detection of renal abnormalities associated with skeletal or brain abnormalities should raise the suspicion for multisystem ciliopathies. Prenatal ultrasound can help to differentiate between different diseases and pave the way for subsequent targeted genetic testing.
A novel heterotaxy gene: Expansion of the phenotype of TTC21B-spectrum disease.
TTC21B encodes the protein IFT139, a critical component of the retrograde transport system within the primary cilium. Biallelic, pathogenic TTC21B variants are associated with classic ciliopathy syndromes, including nephronophthisis, Jeune asphyxiating thoracic dystrophy, and Joubert Syndrome, with ciliopathy-spectrum traits such as biliary dysgenesis, primary ciliary dyskinesia, and situs inversus, and also with focal segmental glomerulosclerosis. We report a 9-year-old male with focal segmental glomerulosclerosis requiring kidney transplant, primary ciliary dyskinesia, and biliary dysgenesis, found by research-based exome sequencing to have biallelic pathogenic TTC21B variants. A sibling with isolated heterotaxy was found to harbor the same variants. This case highlights the phenotypic spectrum and unpredictable manifestations of TTC21B-related disease, and also reports the first association between TTC21B and heterotaxy, nominating TTC21B as an important new heterotaxy gene.
Expression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development.
Joubert syndrome and related disorders (JSRD) and Jeune syndrome are multisystem ciliopathy disorders with overlapping phenotypes. There are a growing number of genetic causes for these rare syndromes, including the recently described genes ARL3 and CEP120. We sought to explore the developmental expression patterns of ARL3 and CEP120 in humans to gain additional understanding of these genetic conditions. We used an RNA in situ detection technique called RNAscope to characterise ARL3 and CEP120 expression patterns in human embryos and foetuses in collaboration with the MRC-Wellcome Trust Human Developmental Biology Resource. Both ARL3 and CEP120 are expressed in early human brain development, including the cerebellum and in the developing retina and kidney, consistent with the clinical phenotypes seen with pathogenic variants in these genes. This study provides insights into the potential pathogenesis of JSRD by uncovering the spatial expression of two JSRD-causative genes during normal human development.
A new case of KIAA0753-related variant of Jeune asphyxiating thoracic dystrophy.
A narrow thorax with shortening of long bones is usually pointing to dysfunction of the primary cilia corresponding clinically to ciliopathies with major skeletal involvement. Mutations in at least 23 genes are likely to correspond to this clinical presentation: IFT43/52/80/81/122/140/172, WDR19/34/35/60, DYNC2H1, DYNC2LI1, CEP120, NEK1, TTC21B, TCTEX1D2, INTU, TCTN3, EVC 1/2 and KIAA0586. In addition to these, KIAA0753 variants were recently described in seven patients with Jeune asphyxiating thoracic dystrophy (ATD) (two first cousins, one unrelated patient and one fetus), Joubert syndrome (two siblings) and orofaciodigital syndrome type 6 (one patient). We present the clinical characteristics of a eighth such patient. This 4 year-old boy with narrow thorax, short limbs, severe respiratory and feeding difficulties from birth on had a history of hypotonia and developmental delay. On skeletal survey, short tubular bones (height - 5,5 SD) and a trident appearance of the pelvis were seen. Brain MRI showed cervical canal stenosis. Renal function was normal and moderate hepatomegaly was noted. A homozygous c.943C > T mutation in KIAA0753 was identified on whole exome sequencing, resulting in Gln315Ter premature termination of the corresponding protein. This case provides confirmation of an additional molecular basis for skeletal dysplasia and illustrates how ciliopathies due to mutations in a single gene may present as apparently distinct syndromes.
CEP120 interacts with C2CD3 and Talpid3 and is required for centriole appendage assembly and ciliogenesis.
Centrosomal protein 120 (CEP120) was originally identified as a daughter centriole-enriched protein that participates in centriole elongation. Recent studies showed that CEP120 gene mutations cause complex ciliopathy phenotypes in humans, including Joubert syndrome and Jeune asphyxiating thoracic dystrophy, suggesting that CEP120 plays an additional role in ciliogenesis. To investigate the potential roles of CEP120 in centriole elongation and cilia formation, we knocked out the CEP120 gene in p53-deficient RPE1 cells using the CRISPR/Cas9 editing system, and performed various analyses. We herein report that loss of CEP120 produces short centrioles with no apparent distal and subdistal appendages. CEP120 knockout was also associated with defective centriole elongation, impaired recruitment of C2CD3 and Talpid3 to the distal ends of centrioles, and consequent defects in centriole appendage assembly and cilia formation. Interestingly, wild-type CEP120 interacts with C2CD3 and Talpid3, whereas a disease-associated CEP120 mutant (I975S) has a low affinity for C2CD3 binding and perturbs cilia assembly. Together, our findings reveal a novel role of CEP120 in ciliogenesis by showing that it interacts with C2CD3 and Talpid3 to assemble centriole appendages and by illuminating the molecular mechanism through which the CEP120 (I975S) mutation causes complex ciliopathies.
Publicações recentes
Mast cell mediators in hereditary angioedema.
Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
🥉 Relato de casoPlatelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
📚 EuropePMC1 artigos no totalmostrando 10
Fetal ciliopathies: a retrospective observational single-center study.
Archives of gynecology and obstetricsA novel heterotaxy gene: Expansion of the phenotype of TTC21B-spectrum disease.
American journal of medical genetics. Part AExpression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development.
BMC developmental biologyA new case of KIAA0753-related variant of Jeune asphyxiating thoracic dystrophy.
European journal of medical geneticsCEP120 interacts with C2CD3 and Talpid3 and is required for centriole appendage assembly and ciliogenesis.
Scientific reportsDisease-Associated Mutations in CEP120 Destabilize the Protein and Impair Ciliogenesis.
Cell reportsA novel Cep120-dependent mechanism inhibits centriole maturation in quiescent cells.
eLifeMutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes.
Journal of medical geneticsMutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes.
Journal of medical geneticsAn siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes.
Nature cell biologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de Joubert com distrofia torácica asfixiante de Jeune.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de Joubert com distrofia torácica asfixiante de Jeune
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Fetal ciliopathies: a retrospective observational single-center study.
- A novel heterotaxy gene: Expansion of the phenotype of TTC21B-spectrum disease.
- Expression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development.
- A new case of KIAA0753-related variant of Jeune asphyxiating thoracic dystrophy.
- CEP120 interacts with C2CD3 and Talpid3 and is required for centriole appendage assembly and ciliogenesis.
- Mast cell mediators in hereditary angioedema.
- Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
- Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
- The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
- Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:397715(Orphanet)
- MONDO:0018342(MONDO)
- GARD:17637(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55346036(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
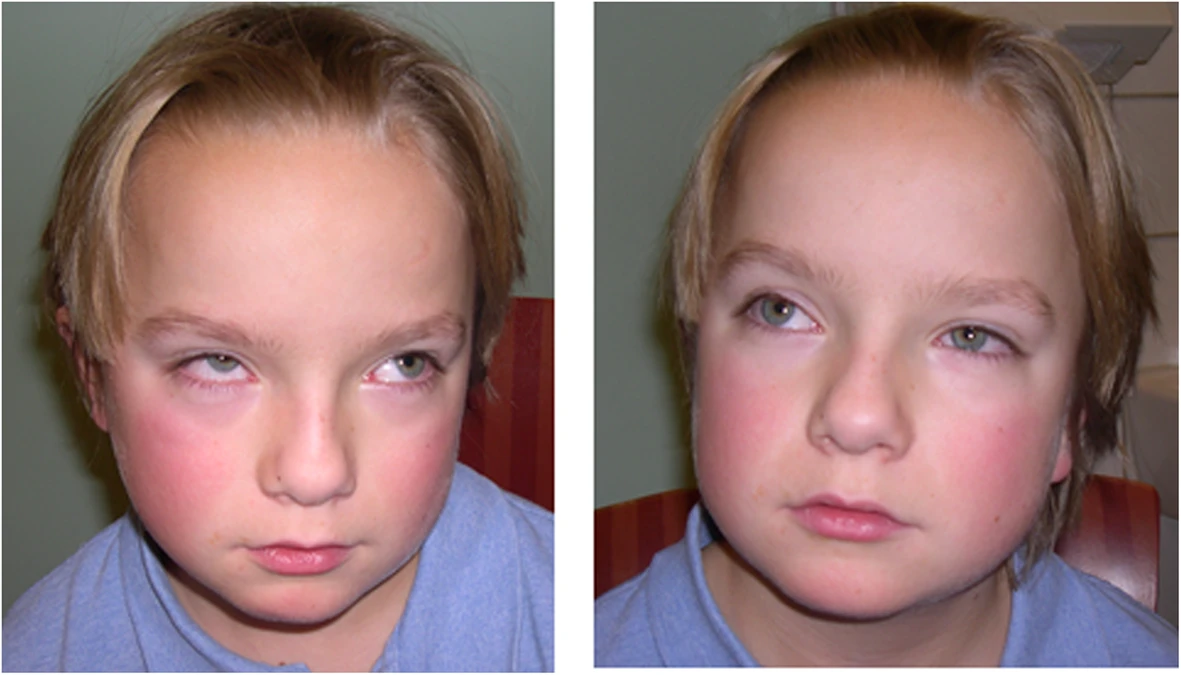
Síndrome de Joubert com distrofia torácica asfixiante de Jeune
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata