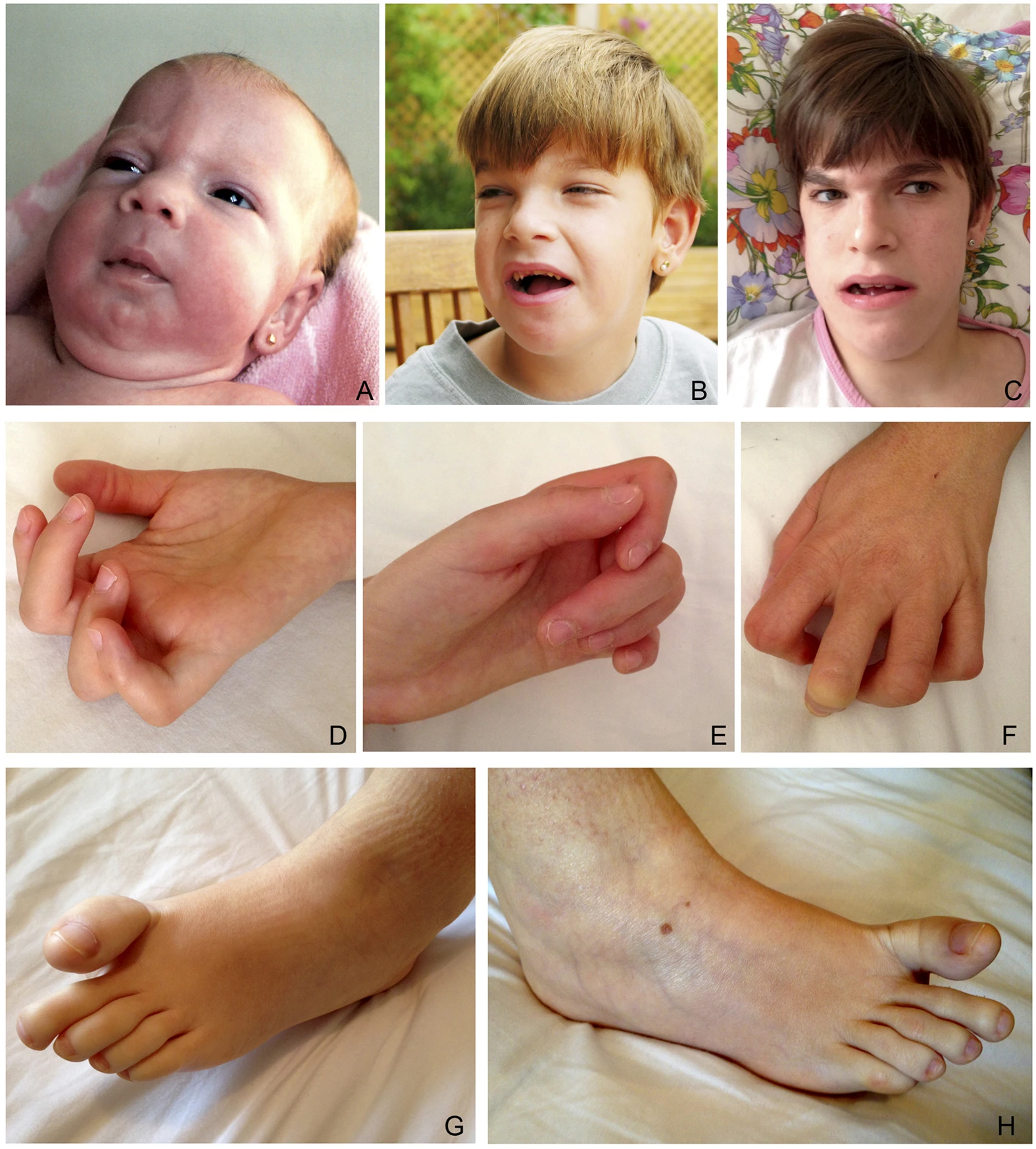
A síndrome de Schaaf-Yang (SYS) é um distúrbio genético raro causado por uma mutação heterozigótica no gene de expressão paterna MAGEL2. Os principais sinais deste distúrbio são: deficiência intelectual/atraso no desenvolvimento, transtorno do espectro autista, hipogonadismo, hipotonia na infância com problemas de alimentação e artrogripose distal. As características faciais incluem nariz curto, sobrancelhas densas e mandíbula proeminente.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome Schaaf-Yang é uma doença genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. O início das manifestações pode ocorrer ainda no período pré-natal (antenatal) ou logo após o nascimento (neonatal). A herança não se aplica aos padrões clássicos, sendo geralmente associada a mutações de novo no gene MAGEL2.[1][4]
Sinais e sintomas
A síndrome apresenta um amplo espectro de manifestações clínicas, que podem incluir: atraso global do desenvolvimento, dificuldade ou incapacidade de andar, fala ausente ou muito limitada, e déficit de crescimento na infância. Características faciais típicas incluem testa estreita, bossas frontais, sobrancelhas espessas, retrognatia (mandíbula recuada), prognatismo mandibular (mandíbula proeminente), boca aberta, filtro nasal anormal, orelhas de implantação baixa e traços faciais grosseiros. Alterações nos membros são comuns, como braquidactilia (dedos curtos), clinodactilia (dedos curvados), camptodactilia (dedos fletidos), dedos afilados, palmas das mãos estreitas e pé em mata-borrão. Podem ocorrer artrogripose múltipla congênita (contraturas articulares), sequência de acinesia fetal (falta de movimentos do feto), instabilidade de temperatura, constipação e obesidade.[1][4]
Causas genéticas
A Síndrome Schaaf-Yang é causada por variantes (mutações) no gene MAGEL2 (MAGE-like protein 2). Este gene está localizado no cromossomo 15 e codifica uma proteína envolvida em processos celulares importantes, como a regulação da transcrição gênica e a sinalização hormonal. As mutações geralmente ocorrem de forma esporádica (de novo), não sendo herdadas dos pais.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica e confirmado por testes genéticos. Os exames disponíveis no Sistema Único de Saúde (SUS) incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); e dosagem de alfa-fetoproteína. O atendimento em reabilitação para doenças raras também está previsto. Atualmente, há 21 testes genéticos disponíveis e 614 variantes registradas no ClinVar para esta condição.[1][2][5][6]
Tratamento e manejo
Não há cura para a Síndrome Schaaf-Yang. O manejo é multidisciplinar e sintomático, visando melhorar a qualidade de vida e o desenvolvimento. O acompanhamento deve incluir fisioterapia, terapia ocupacional, fonoaudiologia e suporte educacional. O tratamento da constipação, da obesidade e da instabilidade de temperatura pode ser necessário. O atendimento em reabilitação para doenças raras está disponível no SUS. Não há medicamentos aprovados especificamente para a síndrome.[1][2][6]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre a síndrome e algumas substâncias, mas estas não constituem recomendações de tratamento. São elas: Glutamina (1 publicação), 2-toluenossulfonamida (1 publicação), Oxigênio (1 publicação) e Sirolimo (1 publicação). Estas informações são provenientes de mineração de textos e não devem ser interpretadas como orientação clínica.[6]
Prognóstico e qualidade de vida
O prognóstico é variável, dependendo da gravidade das manifestações. O atraso global do desenvolvimento e a dificuldade de locomoção e fala podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para maximizar o potencial de desenvolvimento e o bem-estar do paciente.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A síndrome de Schaaf-Yang (SYS) é um distúrbio genético raro causado por uma mutação heterozigótica no gene de expressão paterna MAGEL2. Os principais sinais deste distúrbio são: deficiência intelectual/atraso no desenvolvimento, transtorno do espectro autista, hipogonadismo, hipotonia na infância com problemas de alimentação e artrogripose distal. As características faciais incluem nariz curto, sobrancelhas densas e mandíbula proeminente.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome Schaaf-Yang é uma doença genética rara, com prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. O início das manifestações pode ocorrer ainda no período pré-natal (antenatal) ou logo após o nascimento (neonatal). A herança não se aplica aos padrões clássicos, sendo geralmente associada a mutações de novo no gene MAGEL2.[1][4]
Sinais e sintomas
A síndrome apresenta um amplo espectro de manifestações clínicas, que podem incluir: atraso global do desenvolvimento, dificuldade ou incapacidade de andar, fala ausente ou muito limitada, e déficit de crescimento na infância. Características faciais típicas incluem testa estreita, bossas frontais, sobrancelhas espessas, retrognatia (mandíbula recuada), prognatismo mandibular (mandíbula proeminente), boca aberta, filtro nasal anormal, orelhas de implantação baixa e traços faciais grosseiros. Alterações nos membros são comuns, como braquidactilia (dedos curtos), clinodactilia (dedos curvados), camptodactilia (dedos fletidos), dedos afilados, palmas das mãos estreitas e pé em mata-borrão. Podem ocorrer artrogripose múltipla congênita (contraturas articulares), sequência de acinesia fetal (falta de movimentos do feto), instabilidade de temperatura, constipação e obesidade.[1][4]
Causas genéticas
A Síndrome Schaaf-Yang é causada por variantes (mutações) no gene MAGEL2 (MAGE-like protein 2). Este gene está localizado no cromossomo 15 e codifica uma proteína envolvida em processos celulares importantes, como a regulação da transcrição gênica e a sinalização hormonal. As mutações geralmente ocorrem de forma esporádica (de novo), não sendo herdadas dos pais.[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica e confirmado por testes genéticos. Os exames disponíveis no Sistema Único de Saúde (SUS) incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); e dosagem de alfa-fetoproteína. O atendimento em reabilitação para doenças raras também está previsto. Atualmente, há 21 testes genéticos disponíveis e 614 variantes registradas no ClinVar para esta condição.[1][2][5][6]
Tratamento e manejo
Não há cura para a Síndrome Schaaf-Yang. O manejo é multidisciplinar e sintomático, visando melhorar a qualidade de vida e o desenvolvimento. O acompanhamento deve incluir fisioterapia, terapia ocupacional, fonoaudiologia e suporte educacional. O tratamento da constipação, da obesidade e da instabilidade de temperatura pode ser necessário. O atendimento em reabilitação para doenças raras está disponível no SUS. Não há medicamentos aprovados especificamente para a síndrome.[1][2][6]
Tratamentos citados na literatura
A literatura científica (fonte: PubTator3) menciona associações entre a síndrome e algumas substâncias, mas estas não constituem recomendações de tratamento. São elas: Glutamina (1 publicação), 2-toluenossulfonamida (1 publicação), Oxigênio (1 publicação) e Sirolimo (1 publicação). Estas informações são provenientes de mineração de textos e não devem ser interpretadas como orientação clínica.[6]
Prognóstico e qualidade de vida
O prognóstico é variável, dependendo da gravidade das manifestações. O atraso global do desenvolvimento e a dificuldade de locomoção e fala podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e o suporte multidisciplinar são essenciais para maximizar o potencial de desenvolvimento e o bem-estar do paciente.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 38 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 106 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable.
Curadoria gene-doença
fontes oficiaisProbably enhances ubiquitin ligase activity of RING-type zinc finger-containing E3 ubiquitin-protein ligases, possibly through recruitment and/or stabilization of the Ubl-conjugating enzyme (E2) at the E3:substrate complex. Acts as a regulator of retrograde transport via its interaction with VPS35. Recruited to retromer-containing endosomes and promotes the formation of 'Lys-63'-linked polyubiquitin chains at 'Lys-220' of WASHC1 together with TRIM27, leading to promote endosomal F-actin assembly
Early endosomeCytoplasmNucleus
Schaaf-Yang syndrome
A disease characterized by clinical features of Prader-Willi syndrome, including neonatal hypotonia with poor suck, feeding problems in infancy, obesity, developmental delay, short stature, and hypogonadism. Additionally, patients manifest autism spectrum disorder. Some patients have dysmorphic facial features.
Variantes genéticas (ClinVar)
614 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 160 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Schaaf-Yang
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
The Diagnostic and Therapeutic Challenges of Schaaf-Yang Syndrome: A Brazilian Case Report.
Background: Schaaf-Yang syndrome is an ultra-rare neurodevelopmental disorder caused by pathogenic variants in the MAGEL2 gene, also implicated in Prader-Willi syndrome. With less than 1 case per 50 000 people, Schaaf-Yang syndrome remains underreported globally, and no previous cases have been documented in Brazil. Case description: This study presents a 4-year-old Brazilian girl clinically diagnosed with Prader-Willi syndrome, characterized by hypotonia, delayed neuropsychomotor development, and craniofacial dysmorphism. Initial assessments identified growth hormone deficiency and developmental delays, followed by genetic sequencing at 2 years of age, confirming a pathogenic MAGEL2 variant [NM_019066.5:c2821dup: p (Arg941Profs*10)]. The child was also diagnosed with autism spectrum disorder. Multidisciplinary interventions, including medical care, speech therapy, and psychological support, led to remarkable improvements in motor function, language, memory, and learning. Conclusions: This is the first documented Brazilian case of Schaaf-Yang syndrome, emphasizing the critical role of early diagnosis and personalized interventions in reducing the impact of the syndrome.
Identification of a De Novo MAGEL2 Pathogenic Variant in Schaaf-Yang Syndrome and the Importance of Paternal Allele Confirmation.
Schaaf-Yang syndrome (SYS) is a rare genetic disorder caused by pathogenic variants in MAGEL2, a paternally expressed and maternally imprinted gene on 15q11.2. Genetic diagnosis of SYS is challenging due to clinical features that overlap with those of other rare syndromic disorders and limited clinician awareness regarding the diagnosis of imprinting disorders. We present a neonate presenting with respiratory distress and joint contractures, diagnosed with SYS through comprehensive genetic testing. Whole-exome sequencing identified a heterozygous de novo nonsense variant in MAGEL2 c.2092G>T [p.(Gly698Ter)]. To confirm the paternal origin of this variant, methylation-sensitive restriction enzyme digestion followed by long-range PCR, nested PCR, and Sanger sequencing was performed. The MAGEL2 c.2092G>T was absent in both parents, indicating a de novo mutation. Methylation analysis confirmed that the variant resided on the paternal allele, establishing its pathogenicity. A proband diagnostic approach integrating sequence analysis, parental testing, and methylation assays effectively delineates MAGEL2 variants and their inheritance patterns. Our study underscores the importance of combining genomic sequencing with methylation assay to accurately diagnose SYS, especially for de novo variants. The proposed diagnostic workflow facilitates reliable identification of pathogenic MAGEL2 mutations and their parental origin, improving diagnostic precision in clinical settings. Further research is needed to elucidate the molecular mechanisms underlying MAGEL2-related disorders and develop targeted therapies.
MAGEL2 (patho-)physiology and Schaaf-Yang syndrome.
Schaaf-Yang syndrome (SYS) is a complex neurodevelopmental disorder characterized by autism spectrum disorder, joint contractures, and profound hypothalamic dysfunction. SYS is caused by variants in MAGEL2, a gene within the Prader-Willi syndrome (PWS) locus on chromosome 15. In this review, we consolidate decades of research on MAGEL2 to elucidate its physiological functions. Moreover, we synthesize current knowledge on SYS, suggesting that while MAGEL2 loss-of-function seems to underlie several SYS and PWS phenotypes, additional pathomechanisms probably contribute to the distinct and severe phenotype observed in SYS. In addition, we highlight recent therapeutic advances and identify promising avenues for future investigation.
In-depth behavioral characterization of a rat model of Schaaf-Yang syndrome.
Schaaf-Yang syndrome (SYS, OMIM #615547) is a rare neurodevelopmental disorder caused by truncating variants in the maternally imprinted MAGEL2 gene. It is characterized by intellectual disability, autism spectrum disorder, joint contractures, and feeding difficulties. Although MAGEL2 is deleted in most cases of Prader-Willi syndrome (PWS, OMIM #176270), SYS presents with more severe symptoms, suggesting pathogenic effects of truncated MAGEL2 beyond a mere loss of function. This study expands the behavioral characterization of a novel rat model ("Magel2Pmut rats") which carries a truncating mutation on the paternal allele of Magel2, offering greater construct validity for SYS than previous animal models with Magel2 deletion. While an initial study provided first insights, key domains within the behavioral phenotype of the model remained unexplored. Our comprehensive behavioral analysis, including home-cage monitoring, ultrasonic vocalization analysis, and precise gait assessment, identified several phenotypic alterations potentially relevant to the study of SYS. Magel2Pmut rats exhibited abnormal feeding behavior, changes in early social communication, alterations in gait, aberrant behavior in the elevated plus maze, and delayed decision-making. Additionally, we confirmed that Magel2Pmut rats show phenotypes of abnormal social interaction. These results may reflect core symptoms seen in SYS, underscoring the value of this model for preclinical research on pathophysiology and therapeutics.
Atypical Prader-Willi Syndrome Deletions: Insights Into the Complex Regulation and Phenotypic Variability.
This letter discusses two recent reports of Prader–Willi syndrome (PWS) cases with atypical deletions within the 15q11.2 region. The genetic and clinical complexity highlights the need for caution when interpreting individual cases, supports a symptom‐based management approach, and calls for controlled experimental research to elucidate the regulatory landscape of the PWS locus.
Publicações recentes
Case Report: Schaaf-Yang Syndrome Milder Phenotype Due to Potential Pathogenic Novel Missense Variant as an Unusual Cause of Obesity in a Pediatric Patient.
Two siblings with Schaaf-Yang syndrome treated with growth hormone.
Long-read sequencing enables trio-assisted phasing of de novo variants in the imprinted gene MAGEL2.
Identification of a De Novo MAGEL2 Pathogenic Variant in Schaaf-Yang Syndrome and the Importance of Paternal Allele Confirmation.
In-depth behavioral characterization of a rat model of Schaaf-Yang syndrome.
📚 EuropePMC49 artigos no totalmostrando 82
Identification of a De Novo MAGEL2 Pathogenic Variant in Schaaf-Yang Syndrome and the Importance of Paternal Allele Confirmation.
Journal of clinical laboratory analysisIn-depth behavioral characterization of a rat model of Schaaf-Yang syndrome.
Scientific reportsAtypical Prader-Willi Syndrome Deletions: Insights Into the Complex Regulation and Phenotypic Variability.
Molecular genetics & genomic medicineLaparoscopic Treatment of Severe Gastroesophageal Reflux Disease (GERD) in Schaaf-Yang Syndrome: First Report of Toupet Fundoplication.
CureusThe Diagnostic and Therapeutic Challenges of Schaaf-Yang Syndrome: A Brazilian Case Report.
Journal of child neurologyBiallelic variants in SREK1 downregulating SNORD115 and SNORD116 cause a Prader-Willi-like syndrome.
The Journal of clinical investigationA Case of Prader-Willi Syndrome With a Deletion Including MAGEL2 , NDN , and MKRN3 , but Excluding SNRPN and SNORD116.
American journal of medical genetics. Part ARoles of SNORD115 and SNORD116 ncRNA clusters during neuronal differentiation.
Nature communicationsSex differences in MAGEL2 gene promoter methylation in high functioning autism - trends from a pilot study using nanopore Cas9 targeted long read sequencing.
BMC medical genomicsNeuropeptide therapeutics to repress lateral septum neurons that disable sociability in an autism mouse model.
Cell reports. MedicineMAGEL2 (patho)physiology and Schaaf-Yang syndrome.
Developmental medicine and child neurologyMAGEL2 (patho-)physiology and Schaaf-Yang syndrome.
Developmental medicine and child neurologyTruncated variants of MAGEL2 are involved in the etiologies of the Schaaf-Yang and Prader-Willi syndromes.
American journal of human geneticsAcquiring Social Safety Engages Oxytocin Neurons in the Supraoptic Nucleus: Role of Magel2 Deficiency.
NeuroendocrinologySubcellular localisation of truncated MAGEL2 proteins: insight into the molecular pathology of Schaaf-Yang syndrome.
Journal of medical geneticsVariants of the GNAI1 gene manifest as Prader-Willi-like syndrome: Case report with literature review.
Clinical dysmorphologyThe Pivotal Role of Oxytocin's Mechanism of Thermoregulation in Prader-Willi Syndrome, Schaaf-Yang Syndrome, and Autism Spectrum Disorder.
International journal of molecular sciencesMorbidity and mortality in Schaaf-Yang syndrome.
Annals of translational medicine[Clinical characteristics and genetic analysis of a case of infantile Schaaf-Yang syndrome due to a heterozygous variant of MAGEL2 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsHormonal Imbalances in Prader-Willi and Schaaf-Yang Syndromes Imply the Evolution of Specific Regulation of Hypothalamic Neuroendocrine Function in Mammals.
International journal of molecular sciencesCaregiver-based perception of disease burden in Schaaf-Yang syndrome.
Molecular genetics & genomic medicineReport of two cases of Schaaf-Yang syndrome: Same genotype and different phenotype.
Clinical case reportsEarly onset critically ill infants with Schaaf-Yang syndrome: a retrospective study from the China neonatal genomes project and literature review.
Annals of translational medicineSchaaf-Yang Syndrome: Clinical Phenotype and Effects of 4 years of Growth Hormone Treatment.
Hormone research in paediatricsLate-Onset Pyloric Stenosis and Intussusception With Final Diagnosis of Food Proteins' Hypersensitivity in Schaaf-Yang Syndrome: A Case Report.
JPGN reportsOxytocin receptors in the Magel2 mouse model of autism: Specific region, age, sex and oxytocin treatment effects.
Frontiers in neuroscienceTwo new cases with novel pathogenic variants reflecting the clinical diversity of Schaaf-Yang syndrome.
Clinical geneticsPreimplantation Genetic Testing (PGT) and Prenatal Diagnosis of Schaaf-Yang Syndrome: A Report of Three Families and a Research on Genotype-Phenotype Correlations.
Journal of clinical medicineMagel2 truncation alters select behavioral and physiological outcomes in a rat model of Schaaf-Yang syndrome.
Disease models & mechanisms[Clinical and genetic analysis of a child with Schaaf-Yang syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsNeonatal oxytocin gives the tempo of social and feeding behaviors.
Frontiers in molecular neuroscienceAnalysis of the hypothalamic oxytocin system and oxytocin receptor-expressing astrocytes in a mouse model of Prader-Willi syndrome.
Journal of neuroendocrinologyAdvancing in Schaaf-Yang syndrome pathophysiology: from bedside to subcellular analyses of truncated MAGEL2.
Journal of medical geneticsA nationwide survey of Schaaf-Yang syndrome in Japan.
Journal of human geneticsLongitudinal analysis of electroencephalography pattern changes in an infant with Schaaf-Yang syndrome and a novel mutation in melanoma antigen L2 (MAGEL2).
Molecular genetics & genomic medicineSchaaf-Yang Syndrome: A Real Challenge for Prenatal Diagnosis.
CureusEndosomal Recycling Defects and Neurodevelopmental Disorders.
CellsCopeptin: Utility in Paediatric Patients with Hyponatraemia.
Hormone research in paediatricsThe Spectrum of the Prader-Willi-like Pheno- and Genotype: A Review of the Literature.
Endocrine reviewsPatients with PWS and related syndromes display differentially methylated regions involved in neurodevelopmental and nutritional trajectory.
Clinical epigeneticsThe N-terminal domain of the Schaaf-Yang syndrome protein MAGEL2 likely has a role in RNA metabolism.
The Journal of biological chemistryColon Transit Scintigraphy in Schaaf-Yang Syndrome.
Clinical nuclear medicinePhenotypic spectrum and mechanism analysis of Schaff Yang syndrome: A case report on new mutation of MAGEL2 gene.
MedicineExpanding the spectrum of endocrinopathies identified in Schaaf-Yang syndrome - A case report and review of the literature.
European journal of medical geneticsA retrospective analysis of growth hormone therapy in children with Schaaf-Yang syndrome.
Clinical geneticsThe impact of oxytocin on neurite outgrowth and synaptic proteins in Magel2-deficient mice.
Developmental neurobiologyDiagnosis of Schaaf-Yang syndrome in Korean children with developmental delay and hypotonia.
MedicineThe adult phenotype of Schaaf-Yang syndrome.
Orphanet journal of rare diseasesEmerging roles of the MAGE protein family in stress response pathways.
The Journal of biological chemistryPolysomnographic characteristics and sleep-disordered breathing in Schaaf-Yang syndrome.
Pediatric pulmonologyTwo mouse models carrying truncating mutations in Magel2 show distinct phenotypes.
PloS onePhenotypic spectrum and genetic analysis in the fatal cases of Schaaf-Yang syndrome: Two case reports and literature review.
MedicineA MAGEL2-deubiquitinase complex modulates the ubiquitination of circadian rhythm protein CRY1.
PloS oneA Recurrent Variant in MAGEL2 in Five Siblings with Severe Respiratory Disturbance after Birth.
Molecular syndromologySchaaf-Yang syndrome shows a Prader-Willi syndrome-like phenotype during infancy.
Orphanet journal of rare diseasesmTOR and autophagy pathways are dysregulated in murine and human models of Schaaf-Yang syndrome.
Scientific reportsMutations in MAGEL2 and L1CAM Are Associated With Congenital Hypopituitarism and Arthrogryposis.
The Journal of clinical endocrinology and metabolismCrisponi/cold-induced sweating syndrome: Differential diagnosis, pathogenesis and treatment concepts.
Clinical geneticsMAGEL2-related disorders: A study and case series.
Clinical geneticsGenetic testing and PGD for unexplained recurrent fetal malformations with MAGEL2 gene mutation.
Science China. Life sciencesExome sequencing in Crisponi/cold-induced sweating syndrome-like individuals reveals unpredicted alternative diagnoses.
Clinical genetics[Schaaf-Yang syndrome caused by the new variation of MAGEL2 gene in a case].
Zhonghua er ke za zhi = Chinese journal of pediatricsMagel2 Modulates Bone Remodeling and Mass in Prader-Willi Syndrome by Affecting Oleoyl Serine Levels and Activity.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchNeurocognitive and Neurobehavioral Phenotype of Youth with Schaaf-Yang Syndrome.
Journal of autism and developmental disordersA Novel Mutation of MAGEL2 in a Patient with Schaaf-Yang Syndrome and Hypopituitarism.
International journal of endocrinology and metabolismSchaaf-Yang syndrome overview: Report of 78 individuals.
American journal of medical genetics. Part AThe role of obesity in the fatal outcome of Schaaf-Yang syndrome: Early onset morbid obesity in a patient with a MAGEL2 mutation.
American journal of medical genetics. Part AChronic intestinal pseudo-obstruction syndrome and gastrointestinal malrotation in an infantwith schaaf-yang syndrome - Expanding the phenotypic spectrum.
European journal of medical geneticsChitayat-Hall and Schaaf-Yang syndromes:a common aetiology: expanding the phenotype of MAGEL2-related disorders.
Journal of medical geneticsEarly-onset epileptic encephalopathy and severe developmental delay in an association with de novo double mutations in NF1 and MAGEL2.
Epilepsia openWhole-exome Sequencing Helps the Diagnosis and Treatment in Children with Neurodevelopmental Delay Accompanied Unexplained Dyspnea.
Scientific reportsHormonal, metabolic and skeletal phenotype of Schaaf-Yang syndrome: a comparison to Prader-Willi syndrome.
Journal of medical geneticsPhenotype of two Polish patients with Schaaf-Yang syndrome confirmed by identifying mutation in MAGEL2 gene.
Clinical dysmorphologyThree patients with Schaaf-Yang syndrome exhibiting arthrogryposis and endocrinological abnormalities.
American journal of medical genetics. Part ACellular and disease functions of the Prader-Willi Syndrome gene MAGEL2.
The Biochemical journalMagel2 knockout mice manifest altered social phenotypes and a deficit in preference for social novelty.
Genes, brain, and behaviorA De Novo Nonsense Mutation in MAGEL2 in a Patient Initially Diagnosed as Opitz-C: Similarities Between Schaaf-Yang and Opitz-C Syndromes.
Scientific reportsCORRIGENDUM: The phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families.
Genetics in medicine : official journal of the American College of Medical GeneticsMuscle dysfunction caused by loss of Magel2 in a mouse model of Prader-Willi and Schaaf-Yang syndromes.
Human molecular geneticsThe phenotypic spectrum of Schaaf-Yang syndrome: 18 new affected individuals from 14 families.
Genetics in medicine : official journal of the American College of Medical GeneticsPrader-Willi Syndrome and Schaaf-Yang Syndrome: Neurodevelopmental Diseases Intersecting at the MAGEL2 Gene.
Diseases (Basel, Switzerland)Truncating Mutations of MAGEL2, a Gene within the Prader-Willi Locus, Are Responsible for Severe Arthrogryposis.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Schaaf-Yang.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Schaaf-Yang
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- The Diagnostic and Therapeutic Challenges of Schaaf-Yang Syndrome: A Brazilian Case Report.
- Identification of a De Novo MAGEL2 Pathogenic Variant in Schaaf-Yang Syndrome and the Importance of Paternal Allele Confirmation.
- MAGEL2 (patho-)physiology and Schaaf-Yang syndrome.
- In-depth behavioral characterization of a rat model of Schaaf-Yang syndrome.
- Atypical Prader-Willi Syndrome Deletions: Insights Into the Complex Regulation and Phenotypic Variability.
- Case Report: Schaaf-Yang Syndrome Milder Phenotype Due to Potential Pathogenic Novel Missense Variant as an Unusual Cause of Obesity in a Pediatric Patient.
- Two siblings with Schaaf-Yang syndrome treated with growth hormone.
- Long-read sequencing enables trio-assisted phasing of de novo variants in the imprinted gene MAGEL2.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:398069(Orphanet)
- OMIM OMIM:208080(OMIM)
- MONDO:0014243(MONDO)
- GARD:13316(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q48789662(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Schaaf-Yang
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata