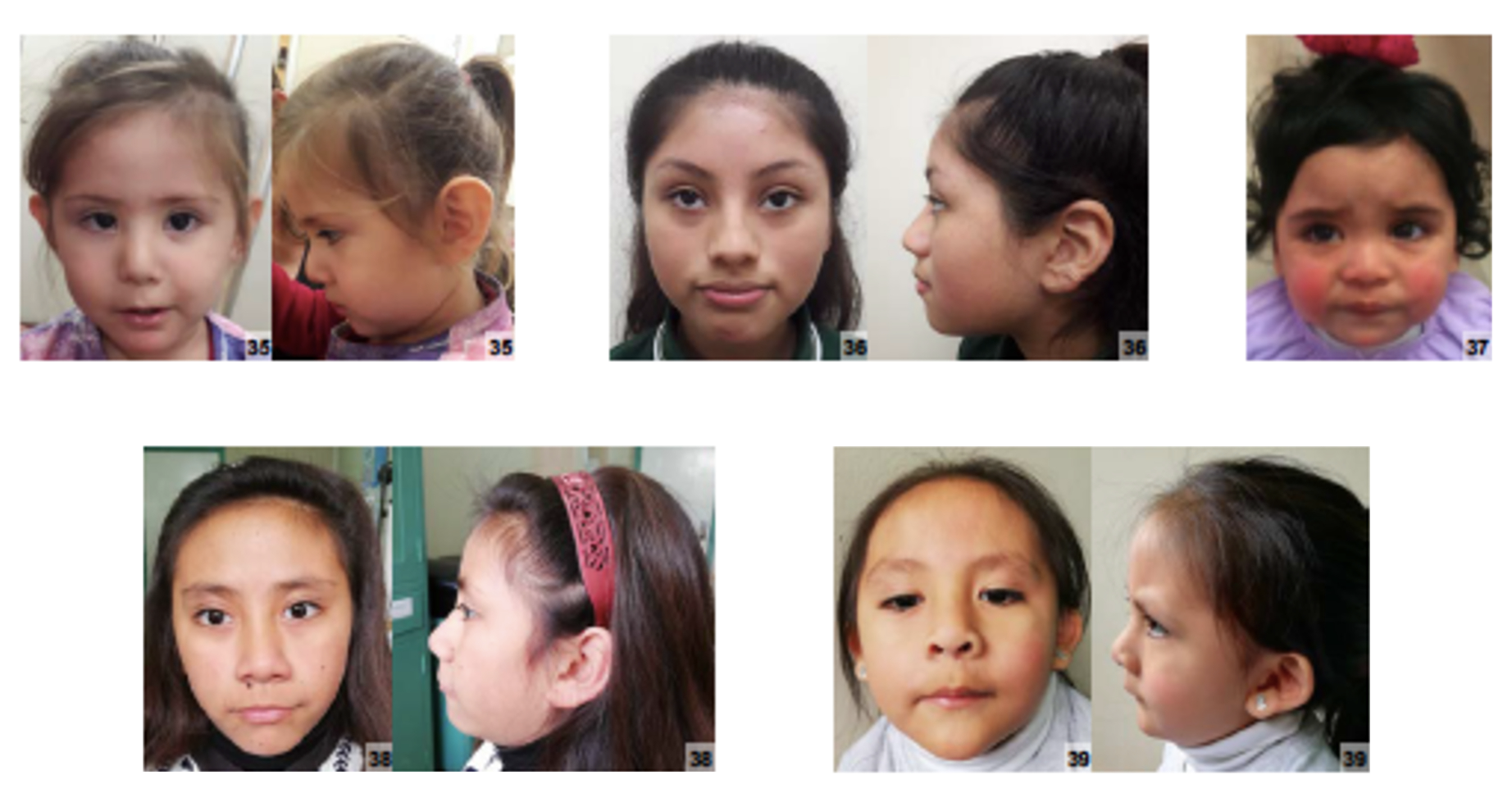
Síndrome neuroocular causada por uma mutação no gene PRR12. Abrange um amplo espectro de anomalias sobrepostas, com atraso no desenvolvimento ou comprometimento do desenvolvimento intelectual como uma descoberta consistente. Anormalidades oculares mostram variabilidade acentuada no tipo e gravidade dos defeitos e incluem anoftalmia, microftalmia e coloboma. Outras características sistêmicas comuns incluem defeitos cardíacos e renais congênitos, hipotonia, retardo de crescimento e microcefalia.
Introdução
O que você precisa saber de cara
Síndrome neuroocular causada por uma mutação no gene PRR12. Abrange um amplo espectro de anomalias sobrepostas, com atraso no desenvolvimento ou comprometimento do desenvolvimento intelectual como uma descoberta consistente. Anormalidades oculares mostram variabilidade acentuada no tipo e gravidade dos defeitos e incluem anoftalmia, microftalmia e coloboma. Outras características sistêmicas comuns incluem defeitos cardíacos e renais congênitos, hipotonia, retardo de crescimento e microcefalia.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 33 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 75 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisMay play a role in the regulation of cohesin complex loading onto chromatin, probably acting in coordination with NIPBL and MAU2
NucleusPostsynaptic densitySynapse, synaptosome
Neuroocular syndrome 1
An autosomal dominant form of neuroocular syndrome, a group of disorders characterized by developmental delay, impaired intellectual development and ocular anomalies as primary findings. Variable eye abnormalities include anophthalmia, microphthalmia, and coloboma. Other common features include congenital heart and kidney defects, hypotonia, failure to thrive, and microcephaly.
Variantes genéticas (ClinVar)
181 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares
Centros de Referência SUS
37 centros habilitados pelo SUS para Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares
Centros para Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
DNA methylation profiling in Kabuki syndrome: reclassification of germline KMT2D VUS and sensitivity in validating postzygotic mosaicism.
Autosomal dominant Kabuki syndrome (KS) is a rare multiple congenital anomalies/neurodevelopmental disorder caused by heterozygous inactivating variants or structural rearrangements of the lysine-specific methyltransferase 2D (KMT2D) gene. While it is often recognizable due to a distinctive gestalt, the disorder is clinically variable, and a phenotypic scoring system has been introduced to help clinicians to reach a clinical diagnosis. The phenotype, however, can be less pronounced in some patients, including those carrying postzygotic mutations. The full spectrum of pathogenic variation in KMT2D has not fully been characterized, which may hamper the clinical classification of a portion of these variants. DNA methylation (DNAm) profiling has successfully been used as a tool to classify variants in genes associated with several neurodevelopmental disorders, including KS. In this work, we applied a KS-specific DNAm signature in a cohort of 13 individuals with KMT2D VUS and clinical features suggestive or overlapping with KS. We succeeded in correctly classifying all the tested individuals, confirming diagnosis for three subjects and rejecting the pathogenic role of 10 VUS in the context of KS. In the latter group, exome sequencing allowed to identify the genetic cause underlying the disorder in three subjects. By testing five individuals with postzygotic pathogenic KMT2D variants, we also provide evidence that DNAm profiling has power to recognize pathogenic variants at different levels of mosaicism, identifying 15% as the minimum threshold for which DNAm profiling can be applied as an informative diagnostic tool in KS mosaics.
A novel missense variant in OTUD5 causes X-linked multiple congenital anomalies-neurodevelopmental syndrome.
The OTUD5 gene encodes a deubiquitinating enzyme (DUB) of the OTU family. Variants of OTUD5 are associated with X-linked multiple congenital anomalies-neurodevelopmental syndrome (MCAND). The case described in this study expands the clinical and molecular spectrum of OTUD5. Trio-based clinical exome sequencing (trio-CES) was performed on a Chinese boy with a clinical phenotype and both of his parents. Sanger sequencing was employed for validation of the variant detected. The patient presented with characteristic facial features, intellectual disability, motor/language/cognitive, and global developmental delays, limb contractures, and kidney abnormalities, and trio-CES identified a de novo missense variant, c.1305T>A, of the OTUD5 gene. We describe OTUD5 gene variation in the Chinese population, with the first report of this variant. Additionally, we provide a comprehensive summary of all published cases of MCAND to date, in order to elucidate the primary clinical features of the syndrome and the variability in phenotype severity. This case expands the genetic and clinical phenotypic spectrum of OTUD5-associated MCAND.
Genetic diagnostic yield in an 11-year cohort of craniosynostosis patients.
Craniosynostosis may present in isolation, 'non-syndromic', or with additional congenital anomalies/neurodevelopmental disorders, 'syndromic'. Clinical focus shifted from confirming classical syndromic cases to offering genetic testing to all craniosynostosis patients. This retrospective study assesses diagnostic yield of molecular testing by investigating prevalences of chromosomal and monogenic (likely) pathogenic variants in an 11-year cohort of 1020 craniosynostosis patients. 502 children underwent genetic testing. Pathogenic variants were identified in 174 patients (35%). Diagnostic yield was significantly higher in syndromic craniosynostosis (62%) than in non-syndromic craniosynostosis (6%). Before whole exome sequencing (WES) emerged, single-gene testing was performed using Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA). Diagnostic yield was 11% and was highest for EFNB1, FGFR2, FGFR3, and IL11RA. Diagnostic yield for copy number variant analysis using microarray was 8%. From 2015 onwards, the WES craniosynostosis panel was implemented, with a yield of 10%. In unsolved, mainly syndromic, cases suspected of a genetic cause, additional WES panels (multiple congenital anomalies (MCA)/intellectual disability (ID)) or open exome analysis were performed with an 18% diagnostic yield. To conclude, microarray and the WES craniosynostosis panel are key to identifying pathogenic variants. in craniosynostosis patients. Given the advances in genetic diagnostics, we should look beyond the scope of the WES craniosynostosis panel and consider extensive genetic diagnostics (e.g. open exome sequencing, whole genome sequencing, RNA sequencing and episignature analysis) if no diagnosis is obtained through microarray and/or WES craniosynostosis panel. If parents are uncomfortable with more extensive diagnostics, MCA or ID panels may be considered.
Publicações recentes
Mast cell mediators in hereditary angioedema.
Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
📚 EuropePMCmostrando 3
DNA methylation profiling in Kabuki syndrome: reclassification of germline KMT2D VUS and sensitivity in validating postzygotic mosaicism.
European journal of human genetics : EJHGA novel missense variant in OTUD5 causes X-linked multiple congenital anomalies-neurodevelopmental syndrome.
Molecular genetics & genomic medicineGenetic diagnostic yield in an 11-year cohort of craniosynostosis patients.
European journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- DNA methylation profiling in Kabuki syndrome: reclassification of germline KMT2D VUS and sensitivity in validating postzygotic mosaicism.
- A novel missense variant in OTUD5 causes X-linked multiple congenital anomalies-neurodevelopmental syndrome.
- Genetic diagnostic yield in an 11-year cohort of craniosynostosis patients.
- Mast cell mediators in hereditary angioedema.
- Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
- Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
- The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
- Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:659904(Orphanet)
- OMIM OMIM:619539(OMIM)
- MONDO:0971007(MONDO)
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de anomalias congênitas múltiplas-transtorno do neurodesenvolvimento-anomalias oculares
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS