Distúrbio metabólico hereditário que afeta a degradação lisossomal dos espingolipídeos. Exemplos representativos incluem a doença de Gaucher, a doença de Tay-Sachs e a doença de Niemann-Pick.
Introdução
O que você precisa saber de cara
Distúrbio metabólico hereditário que afeta a degradação lisossomal dos espingolipídeos. Exemplos representativos incluem a doença de Gaucher, a doença de Tay-Sachs e a doença de Niemann-Pick.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 306 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 760 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
17 genes identificados com associação a esta condição.
Glucosylceramidase that catalyzes, within the lysosomal compartment, the hydrolysis of glucosylceramides/GlcCers (such as beta-D-glucosyl-(1<->1')-N-acylsphing-4-enine) into free ceramides (such as N-acylsphing-4-enine) and glucose (PubMed:15916907, PubMed:24211208, PubMed:32144204, PubMed:39395789, PubMed:9201993). Plays a central role in the degradation of complex lipids and the turnover of cellular membranes (PubMed:27378698). Through the production of ceramides, participates in the PKC-activ
Lysosome membrane
Gaucher disease
An autosomal recessive lysosomal storage disease due to deficient activity of lysosomal beta-glucocerebrosidase, and characterized by accumulation of glucosylceramide in the reticulo-endothelial system. GD is a multisystem disease historically divided into three main subtypes on the basis of the presence of neurologic involvement, age at onset and progression rate: type 1 is the non-neuropathic form, type 2 is the acute neuropathic form with early onset and rapid neurologic deterioration, type 3 is the chronic neuropathic form with slow progression of neurologic features. GD shows a marked phenotypic diversity ranging from adult asymptomatic forms, at the mild end, to perinatal lethal forms at the severe end of the disease spectrum. Formal diagnosis of Gaucher disease is based on the measurement of glucocerebrosidase levels in circulating leukocytes and molecular genetic analysis.
Non-lysosomal glucosylceramidase that catalyzes the hydrolysis of glucosylceramides/GlcCers (such as beta-D-glucosyl-(1<->1')-N-acylsphing-4-enine) to free glucose and ceramides (such as N-acylsphing-4-enine) (PubMed:17105727, PubMed:30308956, PubMed:32144204). GlcCers are membrane glycosphingolipids that have a wide intracellular distribution (By similarity). They are the main precursors of more complex glycosphingolipids that play a role in cellular growth, differentiation, adhesion, signaling
Endoplasmic reticulum membraneGolgi apparatus membrane
Spastic paraplegia 46, autosomal recessive
A neurodegenerative disorder characterized by onset in childhood of slowly progressive spastic paraplegia and cerebellar signs. Some patients have cognitive impairment, cataracts, and cerebral, cerebellar, and corpus callosum atrophy on brain imaging.
Acts as a lysosomal receptor for glucosylceramidase (GBA1) targeting (Microbial infection) Acts as a receptor for enterovirus 71
Lysosome membrane
Epilepsy, progressive myoclonic 4, with or without renal failure
A form of progressive myoclonic epilepsy, a clinically and genetically heterogeneous group of disorders defined by the combination of action and reflex myoclonus, other types of epileptic seizures, and progressive neurodegeneration and neurocognitive impairment. EPM4 is an autosomal recessive form associated with renal failure in some cases. Cognitive function is preserved.
APOE is an apolipoprotein, a protein associating with lipid particles, that mainly functions in lipoprotein-mediated lipid transport between organs via the plasma and interstitial fluids (PubMed:14754908, PubMed:1911868, PubMed:6860692). APOE is a core component of plasma lipoproteins and is involved in their production, conversion and clearance (PubMed:14754908, PubMed:1911868, PubMed:1917954, PubMed:23620513, PubMed:2762297, PubMed:6860692, PubMed:9395455). Apolipoproteins are amphipathic mole
SecretedSecreted, extracellular spaceSecreted, extracellular space, extracellular matrixExtracellular vesicleEndosome, multivesicular body
Hyperlipoproteinemia 3
A disorder characterized by the accumulation of intermediate-density lipoprotein particles (IDL or broad-beta-lipoprotein) rich in cholesterol. Clinical features include xanthomas, yellowish lipid deposits in the palmar crease, or less specific on tendons and on elbows. The disorder rarely manifests before the third decade in men. In women, it is usually expressed only after the menopause.
Lysosomal ceramidase that hydrolyzes sphingolipid ceramides into sphingosine and free fatty acids at acidic pH (PubMed:10610716, PubMed:11451951, PubMed:15655246, PubMed:26898341, PubMed:36752535, PubMed:7744740, PubMed:7852294). Ceramides, sphingosine, and its phosphorylated form sphingosine-1-phosphate are bioactive lipids that mediate cellular signaling pathways regulating several biological processes including cell proliferation, apoptosis and differentiation (PubMed:10610716). Has a higher
LysosomeSecretedNucleusCytoplasm
Farber lipogranulomatosis
An autosomal recessive lysosomal storage disorder characterized by subcutaneous lipid-loaded nodules, excruciating pain in the joints and extremities, and marked accumulation of ceramide in lysosomes. Disease severity is variable. The most severe disease subtype is a rare neonatal form with death occurring before 1 year of age.
ATPase required for the post-translational delivery of tail-anchored (TA) proteins to the endoplasmic reticulum (PubMed:17382883). Recognizes and selectively binds the transmembrane domain of TA proteins in the cytosol. This complex then targets to the endoplasmic reticulum by membrane-bound receptors GET1/WRB and CAMLG/GET2, where the tail-anchored protein is released for insertion. This process is regulated by ATP binding and hydrolysis. ATP binding drives the homodimer towards the closed dime
CytoplasmEndoplasmic reticulumNucleus, nucleolus
Cardiomyopathy, dilated, 2H
A form of dilated cardiomyopathy, a disorder characterized by ventricular dilation and impaired systolic function, resulting in congestive heart failure and arrhythmia. Patients are at risk of premature death. CMD2H is an autosomal recessive form characterized by rapid progression and death in early infancy.
Hydrolyzes the non-reducing end N-acetyl-D-hexosamine and/or sulfated N-acetyl-D-hexosamine of glycoconjugates, such as the oligosaccharide moieties from proteins and neutral glycolipids, or from certain mucopolysaccharides (PubMed:11707436, PubMed:8123671, PubMed:8672428, PubMed:9694901). The isozyme B does not hydrolyze each of these substrates, however hydrolyzes efficiently neutral oligosaccharide (PubMed:11707436). Only the isozyme A is responsible for the degradation of GM2 gangliosides in
LysosomeCytoplasmic vesicle, secretory vesicle, Cortical granule
GM2-gangliosidosis 2
An autosomal recessive lysosomal storage disease marked by the accumulation of GM2 gangliosides in the neuronal cells. Clinically indistinguishable from GM2-gangliosidosis type 1, presenting startle reactions, early blindness, progressive motor and mental deterioration, macrocephaly and cherry-red spots on the macula.
Protein insertase that mediates insertion of transmembrane proteins into the mitochondrial outer membrane (PubMed:36264797). Catalyzes insertion of proteins with alpha-helical transmembrane regions, such as signal-anchored, tail-anchored and multi-pass membrane proteins (By similarity). Does not mediate insertion of beta-barrel transmembrane proteins (By similarity). May play a role in apoptosis (PubMed:12377771)
Mitochondrion outer membrane
The large binding pocket can accommodate several single chain phospholipids and fatty acids, GM2A also exhibits some calcium-independent phospholipase activity (By similarity). Binds gangliosides and stimulates ganglioside GM2 degradation. It stimulates only the breakdown of ganglioside GM2 and glycolipid GA2 by beta-hexosaminidase A. It extracts single GM2 molecules from membranes and presents them in soluble form to beta-hexosaminidase A for cleavage of N-acetyl-D-galactosamine and conversion
Lysosome
GM2-gangliosidosis AB
An autosomal recessive lysosomal storage disease marked by the accumulation of GM2 gangliosides in the neuronal cells. It is characterized by GM2 gangliosides accumulation in the presence of both normal hexosaminidase A and B.
Cleaves beta-linked terminal galactosyl residues from gangliosides, glycoproteins, and glycosaminoglycans Has no beta-galactosidase catalytic activity, but plays functional roles in the formation of extracellular elastic fibers (elastogenesis) and in the development of connective tissue. Seems to be identical to the elastin-binding protein (EBP), a major component of the non-integrin cell surface receptor expressed on fibroblasts, smooth muscle cells, chondroblasts, leukocytes, and certain cance
LysosomeCytoplasm, perinuclear region
GM1-gangliosidosis 1
An autosomal recessive lysosomal storage disease marked by the accumulation of GM1 gangliosides, glycoproteins and keratan sulfate primarily in neurons of the central nervous system. GM1-gangliosidosis type 1 is characterized by onset within the first three months of life, central nervous system degeneration, coarse facial features, hepatosplenomegaly, skeletal dysmorphology reminiscent of Hurler syndrome, and rapidly progressive psychomotor deterioration. Urinary oligosaccharide levels are high. It leads to death usually between the first and second year of life.
Intracellular cholesterol transporter which acts in concert with NPC1 and plays an important role in the egress of cholesterol from the lysosomal compartment (PubMed:11125141, PubMed:15937921, PubMed:17018531, PubMed:18772377, PubMed:29580834). Unesterified cholesterol that has been released from LDLs in the lumen of the late endosomes/lysosomes is transferred by NPC2 to the cholesterol-binding pocket in the N-terminal domain of NPC1 (PubMed:17018531, PubMed:18772377, PubMed:27238017). May bind
SecretedEndoplasmic reticulumLysosome
Niemann-Pick disease C2
A lysosomal storage disorder that affects the viscera and the central nervous system. It is due to defective intracellular processing and transport of low-density lipoprotein derived cholesterol. It causes accumulation of cholesterol in lysosomes, with delayed induction of cholesterol homeostatic reactions. Niemann-Pick disease type C2 has a highly variable clinical phenotype. Clinical features include variable hepatosplenomegaly and severe progressive neurological dysfunction such as ataxia, dystonia and dementia. The age of onset can vary from infancy to late adulthood.
Hydrolyzes the galactose ester bonds of glycolipids such as galactosylceramide and galactosylsphingosine (PubMed:8281145, PubMed:8399327). Enzyme with very low activity responsible for the lysosomal catabolism of galactosylceramide, a major lipid in myelin, kidney and epithelial cells of small intestine and colon (PubMed:8281145, PubMed:8399327)
Lysosome
Krabbe disease
An autosomal recessive disorder characterized by insufficient catabolism of several galactolipids that are important for normal myelin production. Four clinical forms are recognized. The infantile form accounts for 90% of cases. It manifests before six months of age with irritability, spasticity, arrest of motor and mental development, and bouts of temperature elevation without infection. This is followed by myoclonic jerks of arms and legs, oposthotonus, hypertonic fits, and mental regression, which progresses to a severe decerebrate condition with no voluntary movements and death from respiratory infections or cerebral hyperpyrexia before 2 years of age. Cases with later onset present with unexplained blindness, weakness and sensorimotor peripheral neuropathy, mental deterioration and death.
Endoplasmic reticulum (ER)-membrane-bound lysine N-acetyltransferase catalyzing the N6-acetylation of lysine residues in the lumen of the ER in various proteins, including PROM1 and BACE1, using acetyl-CoA as acetyl donor (PubMed:19011241, PubMed:22267734, PubMed:24556617, PubMed:31945187). Thereby, may regulate apoptosis through the acetylation and the regulation of the expression of PROM1 (PubMed:24556617). May also regulate amyloid beta-peptide secretion through acetylation of BACE1 and the r
Endoplasmic reticulum-Golgi intermediate compartment membraneEndoplasmic reticulum membrane
Hydrolyzes the non-reducing end N-acetyl-D-hexosamine and/or sulfated N-acetyl-D-hexosamine of glycoconjugates, such as the oligosaccharide moieties from proteins and neutral glycolipids, or from certain mucopolysaccharides (PubMed:11707436, PubMed:8123671, PubMed:8672428, PubMed:9694901). The isozyme S is as active as the isozyme A on the anionic bis-sulfated glycans, the chondroitin-6-sulfate trisaccharide (C6S-3), and the dermatan sulfate pentasaccharide, and the sulfated glycosphingolipid SM
Lysosome
GM2-gangliosidosis 1
An autosomal recessive lysosomal storage disease marked by the accumulation of GM2 gangliosides in the neuronal cells. It is characterized by GM2 gangliosides accumulation in the absence of HEXA activity, leading to neurodegeneration and, in the infantile form, death in early childhood. It exists in several forms: infantile (most common and most severe), juvenile and adult (late-onset).
Intracellular cholesterol transporter which acts in concert with NPC2 and plays an important role in the egress of cholesterol from the endosomal/lysosomal compartment (PubMed:10821832, PubMed:12554680, PubMed:18772377, PubMed:27238017, PubMed:9211849, PubMed:9927649). Unesterified cholesterol that has been released from LDLs in the lumen of the late endosomes/lysosomes is transferred by NPC2 to the cholesterol-binding pocket in the N-terminal domain of NPC1 (PubMed:18772377, PubMed:19563754, Pu
Late endosome membraneLysosome membrane
Niemann-Pick disease C1
A lysosomal storage disorder that affects the viscera and the central nervous system. It is due to defective intracellular processing and transport of low-density lipoprotein derived cholesterol. It causes accumulation of cholesterol in lysosomes, with delayed induction of cholesterol homeostatic reactions. Niemann-Pick disease type C1 has a highly variable clinical phenotype. Clinical features include variable hepatosplenomegaly and severe progressive neurological dysfunction such as ataxia, dystonia and dementia. The age of onset can vary from infancy to late adulthood. An allelic variant of Niemann-Pick disease type C1 is found in people with Nova Scotia ancestry. Patients with the Nova Scotian clinical variant are less severely affected.
Converts sphingomyelin to ceramide (PubMed:12563314, PubMed:1840600, PubMed:18815062, PubMed:25339683, PubMed:25920558, PubMed:27659707, PubMed:33163980). Exists as two enzymatic forms that arise from alternative trafficking of a single protein precursor, one that is targeted to the endolysosomal compartment, whereas the other is released extracellularly (PubMed:20807762, PubMed:21098024, PubMed:9660788). However, in response to various forms of stress, lysosomal exocytosis may represent a major
LysosomeLipid dropletSecretedSecreted, extracellular space
Niemann-Pick disease A
An early-onset lysosomal storage disorder caused by failure to hydrolyze sphingomyelin to ceramide. It results in the accumulation of sphingomyelin and other metabolically related lipids in reticuloendothelial and other cell types throughout the body, leading to cell death. Niemann-Pick disease type A is a primarily neurodegenerative disorder characterized by onset within the first year of life, intellectual disability, digestive disorders, failure to thrive, major hepatosplenomegaly, and severe neurologic symptoms. The severe neurological disorders and pulmonary infections lead to an early death, often around the age of four. Clinical features are variable. A phenotypic continuum exists between type A (basic neurovisceral) and type B (purely visceral) forms of Niemann-Pick disease, and the intermediate types encompass a cluster of variants combining clinical features of both types A and B.
Oxidase that catalyzes the conversion of cysteine to 3-oxoalanine on target proteins, using molecular oxygen and an unidentified reducing agent (PubMed:12757706, PubMed:15657036, PubMed:15907468, PubMed:16368756, PubMed:21224894, PubMed:25931126). 3-oxoalanine modification, which is also named formylglycine (fGly), occurs in the maturation of arylsulfatases and some alkaline phosphatases that use the hydrated form of 3-oxoalanine as a catalytic nucleophile (PubMed:12757706, PubMed:15657036, PubM
Endoplasmic reticulum lumen
Multiple sulfatase deficiency
A clinically and biochemically heterogeneous disorder caused by the simultaneous impairment of all sulfatases, due to defective post-translational modification and activation. It combines features of individual sulfatase deficiencies such as metachromatic leukodystrophy, mucopolysaccharidosis, chondrodysplasia punctata, hydrocephalus, ichthyosis, neurologic deterioration and developmental delay.
Medicamentos e terapias
Mecanismo: Alpha-galactosidase A stabiliser
Mecanismo: Ceramide glucosyltransferase inhibitor
Mecanismo: Ceramide glucosyltransferase inhibitor
Mecanismo: Programmed cell death protein 1 antagonist
Mecanismo: ARSA exogenous gene
Mecanismo: Ceramide glucosyltransferase inhibitor
Mecanismo: Alpha-galactosidase A stabiliser
Mecanismo: Ceramide glucosyltransferase inhibitor
Variantes genéticas (ClinVar)
627 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
44 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Esfingolipidose
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Brain accumulation of lactosylceramide characterizes GALC deficiency in a zebrafish model of Krabbe disease.
Krabbe disease (KD) is an autosomal recessive sphingolipidosis due to mutations of the GALC gene encoding for the lysosomal β-galactosylceramidase (GALC) that removes β-galactose from β-galactosylceramide, β-lactosylceramide (LacCer) and the neurotoxic metabolite β-galactosylsphingosine (psychosine). At present, the accumulation of psychosine is thought to be the main cause of demyelination, neurodegeneration and neuroinflammation that characterize the early infantile KD with a 1.5-2-year median survival. Currently, the standard of care of KD is haematopoietic stem cell transplantation which, however, improves the lifespan of Krabbe patients only when performed before symptoms appear. Thus, a better understanding of the pathogenesis of KD is required for the development of more efficacious therapeutic approaches. This largely depends upon the availability of novel suitable animal models of the disease. Zebrafish (Danio rerio) represents a useful platform for the study of the mechanisms responsible for human hereditary diseases, including sphingolipidoses, and for the identification of new therapeutics. Two co-orthologues of human GALC have been identified in zebrafish, named galca and galcb. Here, we generated a mutant zebrafish line for each of the two co-orthologues by CRISPR/Cas9 genome editing. Galcb knockout (KO), but not galca KO, exerts a dramatic decrease of total GALC activity both in zebrafish embryos and in the brain of adult mutants. At 3-4 months post-fertilization, galcb KO zebrafish showed impaired locomotion and reduced lifespan. Gene expression analysis, immunohistochemistry, spectral confocal reflectance and transmission electron microscopy showed the presence of demyelination, neuroinflammation and neurodegeneration in the brain of galcb KO mutants. Notably, double galca/galcb KO did not cause a further worsening of the disease when compared with galcb KO mutants. Finally, targeted lipidomic analysis demonstrated a dramatic accumulation of the bioactive sphingolipid LacCer in the brain of both galcb KO and double galca/galcb KO mutants with a modest increase of psychosine levels. Accordingly, activation of LacCer-related signalling occurs in the brain of galcb KO animals. Furthermore, intraventricular injection of LacCer upregulates the expression of various proinflammatory markers and increase mpeg1-positive macrophage infiltration in the brain of 5 dpf zebrafish embryos. In conclusion, galcb KO zebrafish recapitulates several pathological features of KD and is characterized by the accumulation of the bioactive LacCer. This model sheds new light on a possible role of LacCer as a neuroinflammatory/neurodegenerative metabolite in KD with implications for the development of novel therapeutic strategies.
Cytotoxic lymphocyte effector function is unaffected in patients with Gaucher disease.
Gaucher disease (GD) is one of the most common lysosomal storage disorders. It is caused by bi-allelic mutations in the GBA1 gene responsible for the production of β-glucocerebrosidase, an enzyme responsible for the hydrolysis of the sphingolipid glucocerebroside. This results in its accumulation in various organs, and patients can present with a variety of symptoms ranging from visceral enlargement, bone pathology and hematological manifestations. Neuronopathic effects are seen in the severe form of the disease. GD patients also have an increased risk of B-cell malignancies. Some of the hematological symptoms of GD resemble those of the systemic hyperinflammatory condition, hemophagocytic lymphohistiocytosis (HLH). HLH can be familial, due to functional deficiencies in cytotoxic lymphocytes, or acquired from a variety of causes ranging from infections to blood cancers. While patients with inherited and acquired HLH receive the same first-line therapy, patients with the familial form can only be cured by stem cell transplantation, although this treatment may be detrimental to patients with the acquired form of the disease. Therefore, we investigated whether the abnormal lipid accumulation in GD patient cytotoxic lymphocytes and in cells with irreversibly inhibited glucocerebrosidase activity affects their cytotoxicity. Our detailed analysis of primary cytotoxic T lymphocytes and natural killer cells revealed that the activity of these cells was not affected. This finding has important implications for the treatment choices for patients with GD and suggests that these patients can be treated with autologous immunotherapy if they develop hematological cancers.
Therapeutic targeting of neuroinflammation in sphingolipidosis.
Lysosomal storage diseases (LSDs) are a class of hereditary metabolic disorders primarily caused by lysosomal enzyme defects, leading to the accumulation of undegraded substrates. Sphingolipidoses, a subset of LSDs, are primarily associated with profound involvement of the central nervous system (CNS), characterized by progressive neurodegeneration due to massive sphingolipid accumulation. A common pathological feature among many CNS-involved LSDs is the early activation of microglia and astrocytes, which often precedes and predicts regions of subsequent neuronal loss. The extent to which neuroinflammation disrupts CNS homeostasis appears to be determined by its onset, magnitude, and duration. Although neuroinflammatory processes are increasingly recognized as critical contributors to disease progression in sphingolipidoses, the molecular mechanisms underlying glial activation and the initiation of inflammatory cascades remain incompletely understood. Therefore, mouse models of sphingolipidoses have been instrumental in elucidating these pathogenic processes and provide valuable platforms for evaluating therapeutic strategies. This review critically examines the role of neuroinflammation in sphingolipidoses, summarizes insights derived from pre-clinical models, and discusses the therapeutic potential of anti-inflammatory interventions to mitigate CNS pathology and improve clinical outcomes.
Expert review in diagnostic, therapeutic and follow-up of Fabry disease in Latin America based on patient care standards.
Fabry disease (FD) is an X-linked lysosomal sphingolipidosis. It is caused by pathogenic variants in the GLA gene with a consequent deficiency of the enzyme α-galactosidase A, resulting in the pathological accumulation of glycolipids - mainly globotriosyl ceramide (GL-3, GB3) and its deacylated product, globotriaosylsphingosine (Lyso-Gb-3) - in plasma and in a wide variety of cell types throughout the human body; it is characterized as a chronic, multisystemic disease with progressive evolution, which causes deterioration of the patient's quality of life and decreases survival and life expectancy.In Latin America there are different limitations to the management of patients with Fabry disease, in most countries, access to diagnostic tools and treatment on time is complex and can sometimes suffer delays in its implementation. This situation is due to the high costs to health systems of follow-up and pharmacological therapy for Fabry patients, creating barriers to timely access. Although medical criteria are fundamental in the choice of pharmacological therapy, the final decision should also rely on the patient's choice according to their expectations and the adherence and compliance with the treatment that they are willing to follow. As it has been described, there are currently three therapeutic options, for which the appropriate profile must be defined to achieve the best clinical outcomes, considering that it is a permanent treatment; experts consider that Fabry patients need comprehensive and interdisciplinary management to stop the progression and functional deterioration of the affected organs by its multiple systemic manifestations. In Latin-American countries, it is difficult to guarantee this comprehensive and coordinated management, due to limited public policies related to orphan diseases diagnosis, treatment and follow up.It is considered crucial to structure support networks specialized in Fabry disease and generate partnerships with health institutions and other health system stakeholders, that would articulate and coordinate patients and relatives counseling and management, establish the specific pharmacological treatment to reduce the progression of the disease and the systemic involvement, deciding between the administration of enzyme replacement therapy or the most recent option of oral management with pharmacological chaperone both with proven effectiveness. This will be the decision of the attending physician, who will propose and advise the therapeutic choice that best suits the patient's needs.
Evaluation of Lysosphingolipid Analysis for the Diagnosis of Lysosomal Storage Disease.
Lysosomal storage disorders (LSD) are a group of inherited inborn metabolism errors that are characterized by a deficiency in the lysosomal enzyme. In patients with suspected lipid storage disorders, confirmation of the diagnosis relies predominantly on the measurement of specific enzymatic activities and molecular genetic studies. New approaches to the measurement of lysosphingolipids have been developed that may serve as a rapid first-tier screening tests for the evaluation of lysosomal storage disorders. The present study evaluates the results of lysosphingolipid screening tests in patients with suspected lysosomal storage diseases. Lysosphingolipid elevation was detected in five patients examined with suspected lysosomal storage disease, and a definitive diagnosis was reached based on genetic analysis. Our data support recent evidence of the primary role of LysoSLs in the diagnosis of sphingolipidosis, and suggest that these biomarkers may be used for diagnosis and treatment monitoring in the future. Lysosomale Speicherstörungen (LSD) sind eine Gruppe erblicher angeborener Stoffwechselstörungen, die durch einen Mangel des lysosomalen Enzyms gekennzeichnet sind. Bei Patienten mit Verdacht auf Lipidspeicherstörungen stützt sich die Diagnosesicherung überwiegend auf die Messung spezifischer enzymatischer Aktivitäten und molekulargenetische Untersuchungen. Es wurden neue Ansätze zur Messung von Lysosphingolipiden entwickelt, die als schnelle Screening-Tests der ersten Stufe zur Beurteilung lysosomaler Speicherstörungen dienen können. Die vorliegende Studie wertet die Ergebnisse von Lysosphingolipid-Screeningtests bei Patienten mit Verdacht auf lysosomale Speicherkrankheiten aus. Bei fünf untersuchten Patienten mit Verdacht auf eine lysosomale Speicherkrankheit wurde ein Anstieg des Lysosphingolipids festgestellt, und auf der Grundlage einer genetischen Analyse konnte eine endgültige Diagnose gestellt werden. Unsere Daten stützen aktuelle Hinweise auf die primäre Rolle von LysoSLs bei der Diagnose von Sphingolipidose und legen nahe, dass diese Biomarker in Zukunft für die Diagnose und Behandlungsüberwachung verwendet werden könnten.
Publicações recentes
Altered bone structure in Niemann-Pick Type C1 mice, especially in females.
Cytotoxic lymphocyte effector function is unaffected in patients with Gaucher disease.
Therapeutic targeting of neuroinflammation in sphingolipidosis.
Brain accumulation of lactosylceramide characterizes GALC deficiency in a zebrafish model of Krabbe disease.
Expert review in diagnostic, therapeutic and follow-up of Fabry disease in Latin America based on patient care standards.
📚 EuropePMC43 artigos no totalmostrando 63
Cytotoxic lymphocyte effector function is unaffected in patients with Gaucher disease.
Frontiers in immunologyTherapeutic targeting of neuroinflammation in sphingolipidosis.
Molecular immunologyBrain accumulation of lactosylceramide characterizes GALC deficiency in a zebrafish model of Krabbe disease.
Brain : a journal of neurologyExpert review in diagnostic, therapeutic and follow-up of Fabry disease in Latin America based on patient care standards.
Molecular genetics and metabolism reportsEvaluation of Lysosphingolipid Analysis for the Diagnosis of Lysosomal Storage Disease.
Klinische PadiatrieDevelopment of a Multiplexed Sphingolipids Method for Diagnosis of Inborn Errors of Ceramide Metabolism.
Clinical chemistryExpanding the Neurological Phenotype of Anderson-Fabry Disease: Proof of Concept for an Extrapyramidal Neurodegenerative Pattern and Comparison with Monogenic Vascular Parkinsonism.
CellsImpact of an irreversible β-galactosylceramidase inhibitor on the lipid profile of zebrafish embryos.
Computational and structural biotechnology journalImproving newborn screening test performance for metachromatic leukodystrophy: Recommendation from a pre-pilot study that identified a late-infantile case for treatment.
Molecular genetics and metabolismSelective screening for inherited metabolic disorders in a tertiary care hospital of Karachi - A retrospective chart review.
Pakistan journal of medical sciencesAltered Sphingolipid Hydrolase Activities and Alpha-Synuclein Level in Late-Onset Schizophrenia.
Metabolites[Multisystem lesions in orphan diseases: rheumatological aspects of Fabry's disease. Case report].
Terapevticheskii arkhivAnimal Models for the Study of Gaucher Disease.
International journal of molecular sciencesLysoglycosphingolipids have the ability to induce cell death through direct PI3K inhibition.
Journal of neurochemistryThe atypical sphingolipid SPB 18:1(14Z);O2 is a biomarker for DEGS1 related hypomyelinating leukodystrophy.
Journal of lipid researchAlpha-Synuclein mRNA Level Found Dependent on L444P Variant in Carriers and Gaucher Disease Patients on Enzyme Replacement Therapy.
BiomoleculesA zebrafish model of combined saposin deficiency identifies acid sphingomyelinase as a potential therapeutic target.
Disease models & mechanismsImportance to include differential diagnostics for acid sphingomyelinase deficiency (ASMD) in patients suspected to have to Gaucher disease.
Molecular genetics and metabolismGene Therapy of Sphingolipid Metabolic Disorders.
International journal of molecular sciencesPotential Role of Sphingolipidoses-Associated Lysosphingolipids in Cancer.
Cancersβ-Galactosylceramidase Deficiency Causes Upregulation of Long Pentraxin-3 in the Central Nervous System of Krabbe Patients and Twitcher Mice.
International journal of molecular sciencesA rare cause of nephrotic syndrome-sphingosine-1-phosphate lyase (SGPL1) deficiency: 6 cases and a review of the literature.
Pediatric nephrology (Berlin, Germany)Hematological Findings in Lysosomal Storage Disorders: A Perspective from the Medical Laboratory.
EJIFCCA 2-bp deletion mutation in SMPD1 gene leading to lysosomal acid sphingomyelinase deficiency in a Chinese consanguineous pedigree.
Journal of pediatric endocrinology & metabolism : JPEMPlasma neurofilament light, glial fibrillary acidic protein and lysosphingolipid biomarkers for pharmacodynamics and disease monitoring of GM2 and GM1 gangliosidoses patients.
Molecular genetics and metabolism reportsLysosphingolipid urine screening test using mass spectrometry for the early detection of lysosomal storage disorders.
BioanalysisReproduction in Animal Models of Lysosomal Storage Diseases: A Scoping Review.
Frontiers in molecular biosciencesThe Association Between Lysosomal Storage Disorder Genes and Parkinson's Disease: A Large Cohort Study in Chinese Mainland Population.
Frontiers in aging neuroscienceAnalysis of the HEXA, HEXB, ARSA, and SMPD1 Genes in 68 Iranian Patients.
Journal of molecular neuroscience : MNThe incidence rate of hospitalized lysosomal storage diseases in Poland in 2013-2015 based on data from the National Health Fund.
Pediatric endocrinology, diabetes, and metabolismExpression of Ripk1 and DAM genes correlates with severity and progression of Krabbe disease.
Human molecular geneticsExploiting the Potential of Drosophila Models in Lysosomal Storage Disorders: Pathological Mechanisms and Drug Discovery.
BiomedicinesThe role of exosome lipids in central nervous system diseases.
Reviews in the neurosciencesMultiple Sulfatase Deficiency: A Disease Comprising Mucopolysaccharidosis, Sphingolipidosis, and More Caused by a Defect in Posttranslational Modification.
International journal of molecular sciencesResponsiveness of sphingosine phosphate lyase insufficiency syndrome to vitamin B6 cofactor supplementation.
Journal of inherited metabolic diseaseLysosomal Ceramide Metabolism Disorders: Implications in Parkinson's Disease.
Journal of clinical medicineβ-Galactosylceramidase Deficiency Causes Bone Marrow Vascular Defects in an Animal Model of Krabbe Disease.
International journal of molecular sciencesPrimary adrenal insufficiency: New genetic causes and their long-term consequences.
Clinical endocrinologyA novel association between angiokeratoma corporis diffusum and acid sphingomyelinase deficiency.
Pediatric dermatologyClinical findings in Brazilian patients with adult GM1 gangliosidosis.
JIMD reportsDEGS1 variant causes neurological disorder.
European journal of human genetics : EJHGQuantification of 3D Brain Microangioarchitectures in an Animal Model of Krabbe Disease.
International journal of molecular sciencesFabry disease in cardiology practice: Literature review and expert point of view.
Archives of cardiovascular diseasesDefective Sphingolipids Metabolism and Tumor Associated Macrophages as the Possible Links Between Gaucher Disease and Blood Cancer Development.
International journal of molecular sciencesROS Scavenger, Ebselen, Has No Preventive Effect in New Hearing Loss Model Using a Cholesterol-Chelating Agent.
Journal of audiology & otologySGPL1 Deficiency: A Rare Cause of Primary Adrenal Insufficiency.
The Journal of clinical endocrinology and metabolism[Gaucher's disease - an overview about a sphingolipidosis].
Therapeutische Umschau. Revue therapeutiqueCorrelations Between Serum Cholesterol and Vascular Lesions in Fabry Disease Patients.
Circulation journal : official journal of the Japanese Circulation SocietySphingosine phosphate lyase insufficiency syndrome (SPLIS): A novel inborn error of sphingolipid metabolism.
Advances in biological regulationNiemann-Pick type C disease: The atypical sphingolipidosis.
Advances in biological regulationNephrotic syndrome and adrenal insufficiency caused by a variant in SGPL1.
Clinical kidney journalAcid ceramidase deficiency: Farber disease and SMA-PME.
Orphanet journal of rare diseasesPulmonary Involvement in Adult Patients with Inborn Errors of Metabolism.
Respiration; international review of thoracic diseasesDiastereomer-specific quantification of bioactive hexosylceramides from bacteria and mammals.
Journal of lipid researchKrabbe Disease: Report of a Rare Lipid Storage and Neurodegenerative Disorder.
CureusPort-to-port delivery: Mobilization of toxic sphingolipids via extracellular vesicles.
Journal of neuroscience researchLysosphingolipids and sphingolipidoses: Psychosine in Krabbe's disease.
Journal of neuroscience researchThirteen year retrospective review of the spectrum of inborn errors of metabolism presenting in a tertiary center in Saudi Arabia.
Orphanet journal of rare diseasesAnimal models of GM2 gangliosidosis: utility and limitations.
The application of clinical geneticsNiemann-Pick type C: focus on the adolescent/adult onset form.
The International journal of neuroscienceRP-CARS reveals molecular spatial order anomalies in myelin of an animal model of Krabbe disease.
Journal of biophotonicsTreatment of Massive Labial and Gingival Hypertrophy in a Patient With Infantile Systemic Hyalinosis-A Case Report.
Journal of oral and maxillofacial surgery : official journal of the American Association of Oral and Maxillofacial SurgeonsLysosomal Storage Diseases-Regulating Neurodegeneration.
Journal of experimental neuroscienceAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Esfingolipidose.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Esfingolipidose
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Brain accumulation of lactosylceramide characterizes GALC deficiency in a zebrafish model of Krabbe disease.
- Cytotoxic lymphocyte effector function is unaffected in patients with Gaucher disease.
- Therapeutic targeting of neuroinflammation in sphingolipidosis.
- Expert review in diagnostic, therapeutic and follow-up of Fabry disease in Latin America based on patient care standards.
- Evaluation of Lysosphingolipid Analysis for the Diagnosis of Lysosomal Storage Disease.
- Altered bone structure in Niemann-Pick Type C1 mice, especially in females.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79225(Orphanet)
- MONDO:0019255(MONDO)
- GARD:7672(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q2309612(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
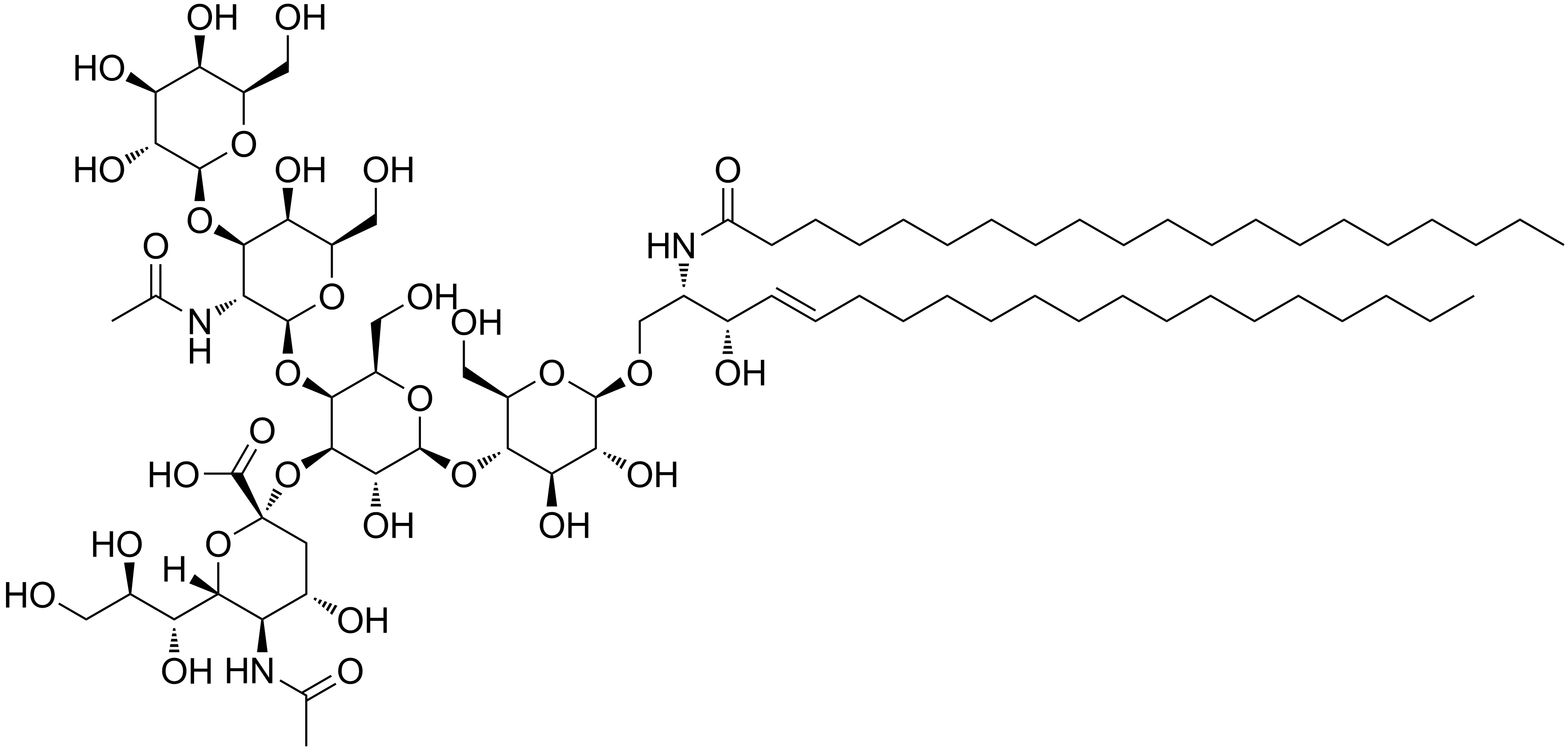
Esfingolipidose
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet