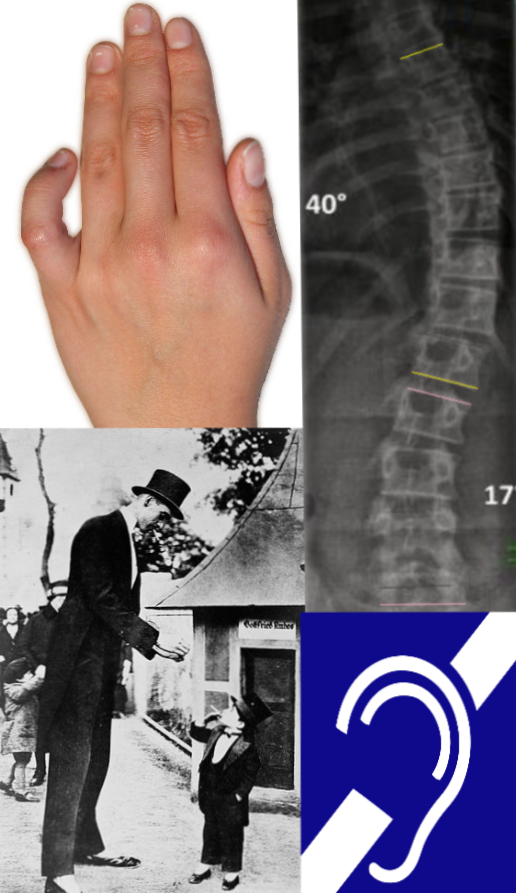
A Síndrome de Camptodactilia, Alta Estatura, Escoliose e Perda Auditiva (CATSHL) é caracterizada por dedos permanentemente dobrados, altura acima da média, curvatura da coluna (escoliose) e dificuldade para ouvir. Ela foi observada em cerca de 30 pessoas de sete gerações da mesma família. A síndrome é causada por uma pequena alteração (mutação de ponto) no gene FGFR3, que faz com que a proteína produzida por esse gene perca parte de sua função. Essa proteína é responsável por controlar o crescimento dos ossos, agindo para diminuí-lo.
Introdução
O que você precisa saber de cara
A Síndrome de Camptodactilia, Alta Estatura, Escoliose e Perda Auditiva (CATSHL) é caracterizada por dedos permanentemente dobrados, altura acima da média, curvatura da coluna (escoliose) e dificuldade para ouvir. Ela foi observada em cerca de 30 pessoas de sete gerações da mesma família. A síndrome é causada por uma pequena alteração (mutação de ponto) no gene FGFR3, que faz com que a proteína produzida por esse gene perca parte de sua função. Essa proteína é responsável por controlar o crescimento dos ossos, agindo para diminuí-lo.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 2 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 19 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Curadoria gene-doença
fontes oficiaisTyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation and apoptosis. Plays an essential role in the regulation of chondrocyte differentiation, proliferation and apoptosis, and is required for normal skeleton development. Regulates both osteogenesis and postnatal bone mineralization by osteoblasts. Promotes apoptosis in chondrocytes, but can also promote cancer cell proliferat
Cell membraneCytoplasmic vesicleEndoplasmic reticulumSecreted
Achondroplasia
A frequent form of short-limb dwarfism. It is characterized by a long, narrow trunk, short extremities, particularly in the proximal (rhizomelic) segments, a large head with frontal bossing, hypoplasia of the midface and a trident configuration of the hands. ACH is an autosomal dominant disease.
Variantes genéticas (ClinVar)
416 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 44 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
16 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de camptodactilia-estatura elevada-escoliose-perda de audição
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
CATSHL syndrome, a new family and phenotypic expansion.
We report the case of a 12-year-old girl and her father who both had marked postnatal tall stature, camptodactyly and clinodactyly, scoliosis and juvenile-onset hearing loss. The CATSHL (CAmptodactyly - Tall stature - Scoliosis - Hearing Loss syndrome) syndrome was suspected, and molecular analysis revealed a hitherto unreported, monoallelic variant c.1861C>T (p.Arg621Cys) in FGFR3. This variant affects the same residue, but is different than, the variant p.Arg621His reported in the two families with dominant CATSHL described so far. Interestingly, peg-shaped incisors were observed in the proband, a feature never reported in CATSHL but typical of another FGFR3-related condition, LADD (Lacrimo - Auricolo - Dento - Digital) syndrome. The FGFR3 p.Arg621Cys variant seems to be a newly identified cause of CATSHL syndrome with some phenotypic overlap with the LADD syndrome.
Fgfr3 mutation disrupts chondrogenesis and bone ossification in zebrafish model mimicking CATSHL syndrome partially via enhanced Wnt/β-catenin signaling.
CATSHL syndrome, characterized by camptodactyly, tall stature and hearing loss, is caused by loss-of-function mutations of fibroblast growth factor receptors 3 (FGFR3) gene. Most manifestations of patients with CATSHL syndrome start to develop in the embryonic stage, such as skeletal overgrowth, craniofacial abnormalities, however, the pathogenesis of these phenotypes especially the early maldevelopment remains incompletely understood. Furthermore, there are no effective therapeutic targets for this skeleton dysplasia. Methods: We generated fgfr3 knockout zebrafish by CRISPR/Cas9 technology to study the developmental mechanisms and therapeutic targets of CATSHL syndrome. Several zebrafish transgenic lines labeling osteoblasts and chondrocytes, and live Alizarin red staining were used to analyze the dynamical skeleton development in fgfr3 mutants. Western blotting, whole mount in situ hybridization, Edu labeling based cell proliferation assay and Wnt/β-catenin signaling antagonist were used to explore the potential mechanisms and therapeutic targets. Results: We found that fgfr3 mutant zebrafish, staring from early development stage, showed craniofacial bone malformation with microcephaly and delayed closure of cranial sutures, chondroma-like lesion and abnormal development of auditory sensory organs, partially resembling the clinical manifestations of patients with CATSHL syndrome. Further studies showed that fgfr3 regulates the patterning and shaping of pharyngeal arches and the timely ossification of craniofacial skeleton. The abnormal development of pharyngeal arch cartilage is related to the augmented hypertrophy and disordered arrangement of chondrocytes, while decreased proliferation, differentiation and mineralization of osteoblasts may be involved in the delayed maturation of skull bones. Furthermore, we revealed that deficiency of fgfr3 leads to enhanced IHH signaling and up-regulated canonical Wnt/β-catenin signaling, and pharmacological inhibition of Wnt/β-catenin could partially alleviate the phenotypes of fgfr3 mutants. Conclusions: Our study further reveals some novel phenotypes and underlying developmental mechanism of CATSHL syndrome, which deepens our understanding of the pathogenesis of CATSHL and the role of fgfr3 in skeleton development. Our findings provide evidence that modulation of Wnt/β-catenin activity could be a potential therapy for CATSHL syndrome and related skeleton diseases.
Publicações recentes
CATSHL syndrome, a new family and phenotypic expansion.
A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de camptodactilia-estatura elevada-escoliose-perda de audição.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de camptodactilia-estatura elevada-escoliose-perda de audição
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- CATSHL syndrome, a new family and phenotypic expansion.
- Fgfr3 mutation disrupts chondrogenesis and bone ossification in zebrafish model mimicking CATSHL syndrome partially via enhanced Wnt/β-catenin signaling.
- A novel mutation in FGFR3 causes camptodactyly, tall stature, and hearing loss (CATSHL) syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:85164(Orphanet)
- OMIM OMIM:610474(OMIM)
- MONDO:0012504(MONDO)
- GARD:10012(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q50349826(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome de camptodactilia-estatura elevada-escoliose-perda de audição
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata