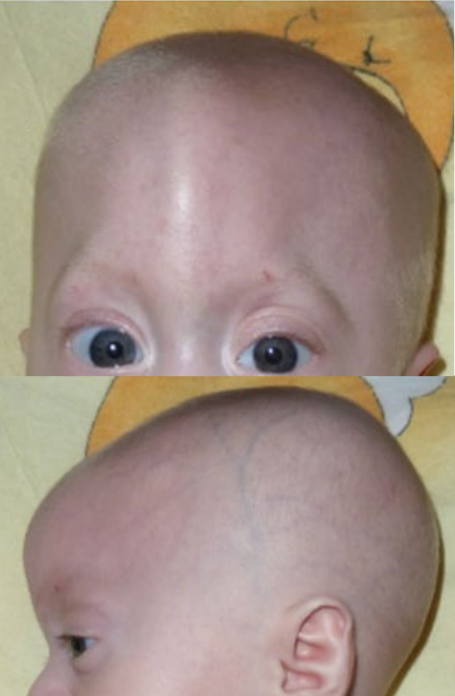
A síndrome de Bohring-Opitz é uma condição rara caracterizada por: * **Crescimento deficiente no útero (IUGR):** o bebê não cresce o suficiente ainda dentro da barriga da mãe. * **Dificuldade para se desenvolver e ganhar peso** após o nascimento. * **Características faciais peculiares,** que incluem: uma linha bem visível na testa (sutura metópica proeminente); uma mancha de nascença avermelhada na testa (nevus flammeus ou "mancha de vinho do porto"); cabelos que nascem muito baixos na testa e nas têmporas, com excesso de pelos (hirsutismo); bochechas cheinhas/inchadas; olhos com o canto externo voltado para cima; olhos um pouco saltados (exoftalmia); olhos mais espaçados do que o normal (hipertelorismo); lábio leporino e fenda no céu da boca; queixo pequeno e para trás (retrognatia); e orelhas em uma posição mais baixa que o comum. * **Deformidades nos cotovelos e punhos,** que ficam curvados. * **Dedos curvados de forma permanente** (camptodactilia). * **Dedos das mãos desviados** para o lado do dedo mínimo (desvio ulnar). * **Alterações nos pés.** * **Atraso grave no desenvolvimento global.** Até o momento, menos de 20 pacientes com essa síndrome foram descritos na literatura médica. Apesar de a maioria dos casos relatados ter surgido de forma isolada (sem uma causa genética clara ou sem que houvesse outros casos na família), também foram registrados casos onde a doença é herdada dos pais de forma recessiva (quando ambos os pais são portadores do gene, mas não manifestam a doença).
Introdução
O que você precisa saber de cara
A síndrome de Bohring-Opitz é uma condição rara caracterizada por: * **Crescimento deficiente no útero (IUGR):** o bebê não cresce o suficiente ainda dentro da barriga da mãe. * **Dificuldade para se desenvolver e ganhar peso** após o nascimento. * **Características faciais peculiares,** que incluem: uma linha bem visível na testa (sutura metópica proeminente); uma mancha de nascença avermelhada na testa (nevus flammeus ou "mancha de vinho do porto"); cabelos que nascem muito baixos na testa e nas têmporas, com excesso de pelos (hirsutismo); bochechas cheinhas/inchadas; olhos com o canto externo voltado para cima; olhos um pouco saltados (exoftalmia); olhos mais espaçados do que o normal (hipertelorismo); lábio leporino e fenda no céu da boca; queixo pequeno e para trás (retrognatia); e orelhas em uma posição mais baixa que o comum. * **Deformidades nos cotovelos e punhos,** que ficam curvados. * **Dedos curvados de forma permanente** (camptodactilia). * **Dedos das mãos desviados** para o lado do dedo mínimo (desvio ulnar). * **Alterações nos pés.** * **Atraso grave no desenvolvimento global.** Até o momento, menos de 20 pacientes com essa síndrome foram descritos na literatura médica. Apesar de a maioria dos casos relatados ter surgido de forma isolada (sem uma causa genética clara ou sem que houvesse outros casos na família), também foram registrados casos onde a doença é herdada dos pais de forma recessiva (quando ambos os pais são portadores do gene, mas não manifestam a doença).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 35 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 116 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisProbable Polycomb group (PcG) protein involved in transcriptional regulation mediated by ligand-bound nuclear hormone receptors, such as retinoic acid receptors (RARs) and peroxisome proliferator-activated receptor gamma (PPARG) (PubMed:16606617). Acts as a coactivator of RARA and RXRA through association with NCOA1 (PubMed:16606617). Acts as a corepressor for PPARG and suppresses its adipocyte differentiation-inducing activity (By similarity). Non-catalytic component of the PR-DUB complex, a co
Nucleus
Bohring-Opitz syndrome
A syndrome characterized by severe intrauterine growth retardation, poor feeding, profound intellectual disability, trigonocephaly, prominent metopic suture, exophthalmos, nevus flammeus of the face, upslanting palpebral fissures, hirsutism, and flexion of the elbows and wrists with deviation of the wrists and metacarpophalangeal joints.
Variantes genéticas (ClinVar)
262 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 170 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome C-like
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Reassessing Benign ASXL1 Variants in Bohring-Opitz Syndrome: The Role of Population Databases in Variant Reinterpretation.
Background/Objectives: ASXL1 is a chromatin-associated gene implicated in both hematologic malignancies and neurodevelopmental disorders, including Bohring-Opitz syndrome (BOS). Although many ASXL1 variants are well classified, a substantial proportion remain variants of uncertain significance (VUS), complicating molecular diagnosis and genetic counseling. The objective of this study was to evaluate whether structural context can inform the interpretation of selected ASXL1 missense variants in a clinical setting. Methods: We describe a 17-year-old female with clinical features consistent with BOS carrying the heterozygous ASXL1 variant p.Q1448R, currently classified as benign under ACMG/AMP guidelines. Three-dimensional in silico structural modeling was performed using AlphaFold3 and available crystallographic data. Three additional ASXL1 missense variants classified as VUS in ClinVar (p.R265H, p.T297M, and p.Y358C) were also analyzed. Evolutionary conservation, domain localization, and residue-level interactions were assessed. Results: Structural modeling indicated that the p.Q1448R substitution alters polar interactions and introduces a steric constraint near a conserved PHD-type zinc finger domain. Variants p.R265H and p.T297M affected stabilizing interactions within the DEUBAD, which is involved in BAP1 activation, while p.Y358C altered a polar microenvironment adjacent to a chromatin-interacting region. All analyzed variants, except p.T297M, localized to evolutionarily conserved regions. Conclusions: This study demonstrates that in silico structural analysis can provide complementary, domain-level insights for the interpretation of ASXL1 missense variants that remain classified as benign, likely benign or VUS under current frameworks. Such approaches may assist in prioritizing variants for further functional evaluation and refining molecular interpretation when experimental data are limited.
Assessing Pubertal Timing, Duration, and Related Characteristics in ASXL-Related Disorders: A Cross-Sectional Caregiver Survey Analysis.
Limited studies have been conducted on pubertal development in populations with pre-existing medical conditions. More than 20-fold increased risk of early puberty has been reported in neurodevelopmental disorders; however, this is a heterogeneous group. There have been limited past studies examining the timing, duration, or characteristics of pubertal or menstrual cycle development in patients with ASXL-related disorders. This study aimed to gather empirical cross-sectional parent survey data regarding pubertal development in adolescents diagnosed with Bohring-Opitz syndrome (BOS) (ASXL1), Shashi-Pena syndrome (SPS) (ASXL2), or Bainbridge-Ropers syndrome (BRS) (ASXL3). Our findings showed evidence for parental and perceived provider concern for premature pubarche and possible precocious puberty (PP) in BOS (ASXL1) males and females. Findings between the BOS (ASXL1) and BRS (ASXL3) individuals differed, representing distinct pubertal phenotypes within these populations. Notable trends toward premature development may warrant a low threshold for pediatric endocrinological evaluation in this population. The characterization and description of a pubertal profile for the ASXL-related syndromes can help inform providers and parents when navigating this stage of development. Our study findings also highlight the need for prospective natural history studies to further define the contribution of pubertal development to the ASXL disorders phenotypes.
Asxl1 loss in mice leads to microcephaly by regulating neural stem cell survival.
Additional sex comb-like 1 (ASXL1) is a chromatin-associated factor essential for transcriptional regulation. De novo truncating mutations in the ASXL1 gene are linked to Bohring-Opitz syndrome, a developmental disorder characterized by microcephaly; however, the role of Asxl1 in brain development remains unclear. In this study, we demonstrate that Asxl1 deletion in mice induces microcephaly, primarily caused by a reduction in the size and number of cortical neurons. Asxl1 ablation disrupts neural stem cell (NSC) maintenance, as evidenced by decreased proliferation and increased apoptosis. Transcriptomic analysis of Asxl1-deficient NSCs revealed 4,635 differentially expressed genes, including 2,262 upregulated and 2,373 downregulated genes. Gene ontology analysis indicated that Asxl1 regulates NSC survival through the histone methyltransferase Ezh2, a core component of the Polycomb Repressive Complex 2 (PRC2). Inhibition of H3K27me3 using GSK343 significantly reduced the viability of wild-type NSCs, but had a markedly diminished effect on Asxl1-deficient NSCs. Furthermore, Ezh2 target genes associated with apoptosis, such as Epha7 and Osr1, were upregulated in wild-type NSCs following GSK343 treatment but not significantly affected in Asxl1-deficient NSCs. These findings establish Asxl1 as a critical regulator of NSC survival and neurogenesis via Ezh2-mediated chromatin modification and provide insights into the mechanisms underlying microcephaly in developmental disorders.
ASXL1 deficiency causes epigenetic dysfunction, combined immunodeficiency, and EBV-associated lymphoma.
Inborn errors of immunity (IEIs) are caused by deleterious variants in immune-related genes. ASXL1 is an epigenetic modifier not previously linked to an IEI. Clonal hematopoiesis and hematologic neoplasms often feature somatic ASXL1 variants, and Bohring-Opitz syndrome, a neurodevelopmental disorder, is caused by heterozygous truncating ASXL1 variants. We present an IEI caused by biallelic germline missense variants in ASXL1. The patient had a history of hematologic abnormalities and viral-associated complications, including chronic macrocytosis, persistent vaccine-strain rubella granulomas, and EBV-associated Hodgkin lymphoma. Immunophenotyping revealed loss of B cells, hypogammaglobulinemia, and impairments in cytotoxic T and NK cell populations. T cells exhibited skewing toward an exhausted memory phenotype, global DNA methylation loss, and increased epigenetic aging. These aberrations were ameliorated by wild-type ASXL1 transduction, confirming the patient variants' pathogenicity. This study defines a novel human IEI caused by ASXL1 deficiency, a diagnosis that should be considered in individuals with chronic viral infections, viral-associated malignancies, and combined immune deficiency.
Bohring-Opitz syndrome: Unraveling neonatal hypoglycemia and early detection through whole exome sequencing.
Publicações recentes
Reassessing Benign ASXL1 Variants in Bohring-Opitz Syndrome: The Role of Population Databases in Variant Reinterpretation.
Assessing Pubertal Timing, Duration, and Related Characteristics in ASXL-Related Disorders: A Cross-Sectional Caregiver Survey Analysis.
ASXL1 deficiency causes epigenetic dysfunction, combined immunodeficiency, and EBV-associated lymphoma.
Asxl1 loss in mice leads to microcephaly by regulating neural stem cell survival.
ASXL1 truncating variants in BOS and myeloid leukemia drive shared disruption of Wnt-signaling pathways but have differential isoform usage of RUNX3.
📚 EuropePMC37 artigos no totalmostrando 50
Reassessing Benign ASXL1 Variants in Bohring-Opitz Syndrome: The Role of Population Databases in Variant Reinterpretation.
GenesAssessing Pubertal Timing, Duration, and Related Characteristics in ASXL-Related Disorders: A Cross-Sectional Caregiver Survey Analysis.
American journal of medical genetics. Part AASXL1 deficiency causes epigenetic dysfunction, combined immunodeficiency, and EBV-associated lymphoma.
The Journal of experimental medicineAsxl1 loss in mice leads to microcephaly by regulating neural stem cell survival.
Animal cells and systemsASXL1 truncating variants in BOS and myeloid leukemia drive shared disruption of Wnt-signaling pathways but have differential isoform usage of RUNX3.
BMC medical genomicsRole of Nasopharyngeal Airway in Management of Craniofacial Syndrome-Associated Upper Airway Obstruction in Children.
Orthodontics & craniofacial researchASXL1-related Bohring-Optiz syndrome complicated by persistent neonatal pulmonary hypertension and abnormal alveoli formation.
European journal of medical geneticsBohring-Opitz syndrome: Unraveling neonatal hypoglycemia and early detection through whole exome sequencing.
Pediatrics and neonatologyTwo cases of hepatoblastoma in Bohring-Opitz syndrome: An emerging association.
Pediatric blood & cancerPerthes-Like Disorder in a Child with Atypical Bohring-Opitz Syndrome.
JBJS case connectorExamining the neurodevelopmental and motor phenotypes of Bohring-Opitz syndrome (ASXL1) and Bainbridge-Ropers syndrome (ASXL3).
Frontiers in neuroscienceMultiomics of Bohring-Opitz syndrome truncating ASXL1 mutations identify canonical and noncanonical Wnt signaling dysregulation.
JCI insightClinical findings in 39 individuals with Bohring-Opitz syndrome from a global patient-driven registry with implications for tumor surveillance and recurrence risk.
American journal of medical genetics. Part ARapid progression of myelofibrosis in polycythemia vera patient carrying SRSF2 c.284C>A p.(Pro95His) and unique ASXL1 splice site c.1720-2A>G variant.
Journal of clinical laboratory analysisDNA methylation signature associated with Bohring-Opitz syndrome: a new tool for functional classification of variants in ASXL genes.
European journal of human genetics : EJHGDe novo nonsense variant in ASXL3 in a Chinese girl causing Bainbridge-Ropers syndrome: A case report and review of literature.
Molecular genetics & genomic medicineBohring-Opitz syndrome caused by a novel ASXL1 mutation (c.3762delT) in an IVF baby: A case report.
MedicineA de novo Variant of ASXL1 Is Associated With an Atypical Phenotype of Bohring-Opitz Syndrome: Case Report and Literature Review.
Frontiers in pediatricsSelf-Induced Bilateral Retinal Detachments and Traumatic Cataracts in a Patient With Bohring-Opitz Syndrome.
Ophthalmic surgery, lasers & imaging retinaAnaesthesia and orphan diseases: Bohring-Opitz syndrome.
European journal of anaesthesiologyUnderstanding the phenotypic spectrum of ASXL-related disease: Ten cases and a review of the literature.
American journal of medical genetics. Part ANovel truncating mutations in ASXL1 identified in two boys with Bohring-Opitz syndrome.
European journal of medical geneticsFurther expanding the clinical phenotype in Bainbridge-Ropers syndrome and dissecting genotype-phenotype correlation in the ASXL3 mutational cluster regions.
European journal of medical geneticsThe tale of two genes: from next-generation sequencing to phenotype.
Cold Spring Harbor molecular case studiesA novel PTC mutation in the BTB domain of KLHL7 gene in two patients with Bohring-Opitz syndrome-like features.
European journal of medical geneticsA de novo truncating mutation in ASXL1 associated with segmental overgrowth.
Journal of geneticsExtending the phenotypic spectrum of Bohring-Opitz syndrome: Mild case confirmed by functional studies.
American journal of medical genetics. Part ADouble outlet right ventricle and aortopulmonary window in a neonate with Bohring-Opitz (Oberklaid-Danks) syndrome: First case report.
Journal of family medicine and primary carePathological ASXL1 Mutations and Protein Variants Impair Neural Crest Development.
Stem cell reportsNew macular findings in individuals with biallelic KLHL7 gene mutation.
BMJ open ophthalmologyTwo siblings with a novel nonsense variant provide further delineation of the spectrum of recessive KLHL7 diseases.
European journal of medical geneticsThe ASXL1 mutation p.Gly646Trpfs*12 found in a Turkish boy with Bohring-Opitz Syndrome.
Clinical case reportsLethal persistent pulmonary hypertension of the newborn in Bohring-Opitz syndrome.
American journal of medical genetics. Part ABohring-Opitz syndrome caused by an ASXL1 mutation inherited from a germline mosaic mother.
American journal of medical genetics. Part ALoss of ASXL1 in the bone marrow niche dysregulates hematopoietic stem and progenitor cell fates.
Cell discoveryA de novo nonsense mutation in ASXL3 shared by siblings with Bainbridge-Ropers syndrome.
Cold Spring Harbor molecular case studiesSyndromic Craniosynostosis Can Define New Candidate Genes for Suture Development or Result from the Non-specifc Effects of Pleiotropic Genes: Rasopathies and Chromatinopathies as Examples.
Frontiers in neuroscienceExpanding the clinical spectrum of recessive truncating mutations of KLHL7 to a Bohring-Opitz-like phenotype.
Journal of medical geneticsASXL gain-of-function truncation mutants: defective and dysregulated forms of a natural ribosomal frameshifting product?
Biology directBohring-opitz syndrome - A case of a rare genetic disorder.
The Medical journal of MalaysiaPathogenic ASXL1 somatic variants in reference databases complicate germline variant interpretation for Bohring-Opitz Syndrome.
Human mutationBainbridge-Ropers syndrome caused by loss-of-function variants in ASXL3: a recognizable condition.
European journal of human genetics : EJHGLoss of Asxl1 Alters Self-Renewal and Cell Fate of Bone Marrow Stromal Cell, Leading to Bohring-Opitz-like Syndrome in Mice.
Stem cell reportsNovel splicing mutation in the ASXL3 gene causing Bainbridge-Ropers syndrome.
American journal of medical genetics. Part AA novel de-novo frameshift mutation of the ASXL1 gene in a classic case of Bohring-Opitz syndrome.
Clinical dysmorphologyScreening of CD96 and ASXL1 in 11 patients with Opitz C or Bohring-Opitz syndromes.
American journal of medical genetics. Part APenetrance of pathogenic mutations in haploinsufficient genes for intellectual disability and related disorders.
European journal of medical geneticsBohring-Opitz syndrome (BOS) with a new ASXL1 pathogenic variant: Review of the most prevalent molecular and phenotypic features of the syndrome.
American journal of medical genetics. Part ACancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex.
Nature communicationsClinical management of patients with ASXL1 mutations and Bohring-Opitz syndrome, emphasizing the need for Wilms tumor surveillance.
American journal of medical genetics. Part AAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome C-like.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome C-like
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Reassessing Benign ASXL1 Variants in Bohring-Opitz Syndrome: The Role of Population Databases in Variant Reinterpretation.
- Assessing Pubertal Timing, Duration, and Related Characteristics in ASXL-Related Disorders: A Cross-Sectional Caregiver Survey Analysis.
- Asxl1 loss in mice leads to microcephaly by regulating neural stem cell survival.
- ASXL1 deficiency causes epigenetic dysfunction, combined immunodeficiency, and EBV-associated lymphoma.
- Bohring-Opitz syndrome: Unraveling neonatal hypoglycemia and early detection through whole exome sequencing.
- ASXL1 truncating variants in BOS and myeloid leukemia drive shared disruption of Wnt-signaling pathways but have differential isoform usage of RUNX3.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:97297(Orphanet)
- OMIM OMIM:605039(OMIM)
- MONDO:0011510(MONDO)
- GARD:10140(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4938225(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome C-like
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov