Disostose acrofacial é um distúrbio genético raro caracterizado por anomalias craniofaciais, como retração da face média e disostose mandibulofacial, além de malformações nos membros e vértebras. Pode apresentar traqueobroncomalácia e necessitar de gastrostomia na infância.
Introdução
O que você precisa saber de cara
Visão geral
A Disostose acrofacial é uma condição genética rara que afeta o desenvolvimento dos ossos da face (crânio e mandíbula) e dos membros (mãos e pés). A palavra "acrofacial" combina "acro" (relativo às extremidades) e "facial" (relativo à face), indicando justamente as duas principais áreas do corpo envolvidas. A condição faz parte de um grupo de doenças conhecidas como disostoses, que são caracterizadas por malformações ósseas. Por ser uma condição rara, sua prevalência exata na população não é conhecida.[1][3]
Sinais e sintomas
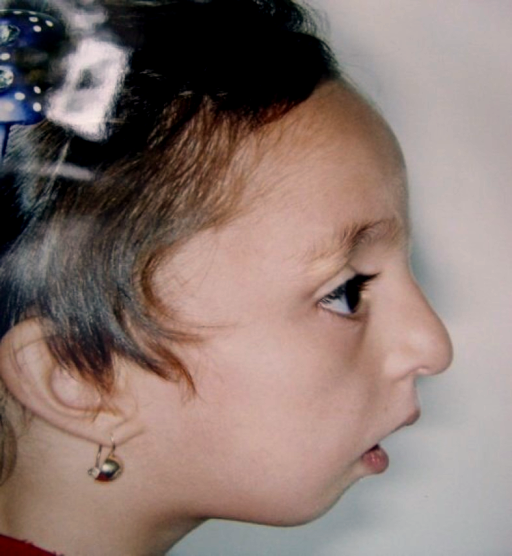
Os sinais e sintomas da Disostose acrofacial podem variar amplamente de uma pessoa para outra, mas geralmente envolvem alterações na face e nos membros. As características faciais mais comuns incluem: retrusão médio-facial (parte central do rosto mais recuada), disostose mandibulofacial (desenvolvimento anormal dos ossos da face e mandíbula), morfologia anormal da pálpebra, sobrancelha fina, anormalidade da glabela (região entre as sobrancelhas), asas nasais espessas e deslocamento pré-auricular do cabelo (cabelo que nasce na frente da orelha). Problemas dentários como agenesia dentária (falta de dentes) e erupção dentária avançada também podem ocorrer.[1][3]
Nas extremidades, os sintomas podem incluir: polegar largo, focomelia (encurtamento ou ausência de partes dos membros), displasia esquelética (desenvolvimento anormal dos ossos), forma anormal dos corpos vertebrais, anormalidade da morfologia da epífise (extremidades dos ossos longos) e displasia da unha do pé. Outros sinais menos frequentes, mas possíveis, são: fissura labial unilateral (lábio leporino), microcefalia secundária (cabeça menor que o esperado, que surge após o nascimento), traqueobroncomalácia (enfraquecimento das vias aéreas), defeito parcial do canal atrioventricular (um tipo de cardiopatia congênita), valva aórtica bicúspide (alteração na válvula do coração), megacólon agangliônico (Doença de Hirschsprung), anormalidade da bigorna e morfologia anormal dos ossículos da orelha média (que podem afetar a audição), calcificação da foice do cérebro e necessidade de alimentação por gastrostomia na infância.[1][3]
Causas genéticas
A Disostose acrofacial é uma condição genética, o que significa que é causada por alterações (mutações) em genes específicos. Vários genes já foram associados a esta condição, incluindo: SF3B4, EVC2, EFTUD2, ZSWIM6, DHODH, EVC, POLR1A e CTNNB1. Cada um desses genes desempenha um papel importante no desenvolvimento do corpo, especialmente na formação dos ossos e cartilagens. A herança (padrão de transmissão familiar) da condição não está claramente definida, podendo variar de acordo com o gene envolvido.[1][4]
Diagnóstico
O diagnóstico da Disostose acrofacial é baseado na avaliação clínica dos sinais e sintomas característicos, especialmente as alterações faciais e dos membros. Para confirmar o diagnóstico, o exame padrão-ouro é o teste genético molecular, que pode identificar mutações em um dos genes associados à doença. Atualmente, existem 336 testes genéticos disponíveis para esta condição, e mais de 840 variantes genéticas diferentes já foram registradas no ClinVar, um banco de dados internacional de variantes genéticas. O código da doença no MONDO (ontologia de doenças) é MONDO:0018237.[1][4][5]
Tratamento e manejo
Não existe um tratamento curativo específico para a Disostose acrofacial. O manejo da condição é multidisciplinar e focado no tratamento dos sintomas e na melhora da qualidade de vida. As abordagens podem incluir cirurgias corretivas para as malformações faciais (como fissura labial) e dos membros, acompanhamento com cardiologista para as cardiopatias congênitas, suporte nutricional (incluindo gastrostomia, se necessário), fisioterapia e terapia ocupacional. O tratamento deve ser individualizado, de acordo com as necessidades de cada paciente. Não há medicamentos aprovados especificamente para a doença. No Brasil, a condição não possui cobertura específica pelo Sistema Único de Saúde (SUS) para procedimentos ou medicamentos.[1][5]
Tratamentos citados na literatura
A literatura científica, através de mineração de dados (PubTator3), menciona algumas substâncias em associação com a Disostose acrofacial. É importante destacar que estas são apenas associações encontradas em publicações científicas e NÃO representam recomendações de tratamento. As substâncias e o número de publicações associadas são: Orotic Acid (2 publicações), pyrimidine (2 publicações), 4 5 dihydroorotic acid (1 publicação), Calcium (1 publicação), Copper (1 publicação), Galactosylceramides (1 publicação), Lipids (1 publicação), Polytetrafluoroethylene (1 publicação), Psychosine (1 publicação) e Silver (1 publicação). Consulte sempre seu médico antes de considerar qualquer intervenção.[5]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Disostose acrofacial é variável e depende da gravidade dos sintomas e da presença de malformações associadas, especialmente cardíacas e respiratórias. Com um manejo multidisciplinar adequado, muitas pessoas podem ter uma qualidade de vida razoável. O acompanhamento regular com uma equipe de especialistas (geneticista, pediatra, cirurgião, cardiologista, fonoaudiólogo, entre outros) é fundamental para monitorar o desenvolvimento e tratar precocemente as complicações. Não há dados disponíveis sobre a expectativa de vida específica para esta condição.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Disostose acrofacial é um distúrbio genético raro caracterizado por anomalias craniofaciais, como retração da face média e disostose mandibulofacial, além de malformações nos membros e vértebras. Pode apresentar traqueobroncomalácia e necessitar de gastrostomia na infância.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Disostose acrofacial é uma condição genética rara que afeta o desenvolvimento dos ossos da face (crânio e mandíbula) e dos membros (mãos e pés). A palavra "acrofacial" combina "acro" (relativo às extremidades) e "facial" (relativo à face), indicando justamente as duas principais áreas do corpo envolvidas. A condição faz parte de um grupo de doenças conhecidas como disostoses, que são caracterizadas por malformações ósseas. Por ser uma condição rara, sua prevalência exata na população não é conhecida.[1][3]
Sinais e sintomas
Os sinais e sintomas da Disostose acrofacial podem variar amplamente de uma pessoa para outra, mas geralmente envolvem alterações na face e nos membros. As características faciais mais comuns incluem: retrusão médio-facial (parte central do rosto mais recuada), disostose mandibulofacial (desenvolvimento anormal dos ossos da face e mandíbula), morfologia anormal da pálpebra, sobrancelha fina, anormalidade da glabela (região entre as sobrancelhas), asas nasais espessas e deslocamento pré-auricular do cabelo (cabelo que nasce na frente da orelha). Problemas dentários como agenesia dentária (falta de dentes) e erupção dentária avançada também podem ocorrer.[1][3]
Nas extremidades, os sintomas podem incluir: polegar largo, focomelia (encurtamento ou ausência de partes dos membros), displasia esquelética (desenvolvimento anormal dos ossos), forma anormal dos corpos vertebrais, anormalidade da morfologia da epífise (extremidades dos ossos longos) e displasia da unha do pé. Outros sinais menos frequentes, mas possíveis, são: fissura labial unilateral (lábio leporino), microcefalia secundária (cabeça menor que o esperado, que surge após o nascimento), traqueobroncomalácia (enfraquecimento das vias aéreas), defeito parcial do canal atrioventricular (um tipo de cardiopatia congênita), valva aórtica bicúspide (alteração na válvula do coração), megacólon agangliônico (Doença de Hirschsprung), anormalidade da bigorna e morfologia anormal dos ossículos da orelha média (que podem afetar a audição), calcificação da foice do cérebro e necessidade de alimentação por gastrostomia na infância.[1][3]
Causas genéticas
A Disostose acrofacial é uma condição genética, o que significa que é causada por alterações (mutações) em genes específicos. Vários genes já foram associados a esta condição, incluindo: SF3B4, EVC2, EFTUD2, ZSWIM6, DHODH, EVC, POLR1A e CTNNB1. Cada um desses genes desempenha um papel importante no desenvolvimento do corpo, especialmente na formação dos ossos e cartilagens. A herança (padrão de transmissão familiar) da condição não está claramente definida, podendo variar de acordo com o gene envolvido.[1][4]
Diagnóstico
O diagnóstico da Disostose acrofacial é baseado na avaliação clínica dos sinais e sintomas característicos, especialmente as alterações faciais e dos membros. Para confirmar o diagnóstico, o exame padrão-ouro é o teste genético molecular, que pode identificar mutações em um dos genes associados à doença. Atualmente, existem 336 testes genéticos disponíveis para esta condição, e mais de 840 variantes genéticas diferentes já foram registradas no ClinVar, um banco de dados internacional de variantes genéticas. O código da doença no MONDO (ontologia de doenças) é MONDO:0018237.[1][4][5]
Tratamento e manejo
Não existe um tratamento curativo específico para a Disostose acrofacial. O manejo da condição é multidisciplinar e focado no tratamento dos sintomas e na melhora da qualidade de vida. As abordagens podem incluir cirurgias corretivas para as malformações faciais (como fissura labial) e dos membros, acompanhamento com cardiologista para as cardiopatias congênitas, suporte nutricional (incluindo gastrostomia, se necessário), fisioterapia e terapia ocupacional. O tratamento deve ser individualizado, de acordo com as necessidades de cada paciente. Não há medicamentos aprovados especificamente para a doença. No Brasil, a condição não possui cobertura específica pelo Sistema Único de Saúde (SUS) para procedimentos ou medicamentos.[1][5]
Tratamentos citados na literatura
A literatura científica, através de mineração de dados (PubTator3), menciona algumas substâncias em associação com a Disostose acrofacial. É importante destacar que estas são apenas associações encontradas em publicações científicas e NÃO representam recomendações de tratamento. As substâncias e o número de publicações associadas são: Orotic Acid (2 publicações), pyrimidine (2 publicações), 4 5 dihydroorotic acid (1 publicação), Calcium (1 publicação), Copper (1 publicação), Galactosylceramides (1 publicação), Lipids (1 publicação), Polytetrafluoroethylene (1 publicação), Psychosine (1 publicação) e Silver (1 publicação). Consulte sempre seu médico antes de considerar qualquer intervenção.[5]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Disostose acrofacial é variável e depende da gravidade dos sintomas e da presença de malformações associadas, especialmente cardíacas e respiratórias. Com um manejo multidisciplinar adequado, muitas pessoas podem ter uma qualidade de vida razoável. O acompanhamento regular com uma equipe de especialistas (geneticista, pediatra, cirurgião, cardiologista, fonoaudiólogo, entre outros) é fundamental para monitorar o desenvolvimento e tratar precocemente as complicações. Não há dados disponíveis sobre a expectativa de vida específica para esta condição.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 140 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 396 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
8 genes identificados com associação a esta condição.
Component of the 17S U2 SnRNP complex of the spliceosome, a large ribonucleoprotein complex that removes introns from transcribed pre-mRNAs (PubMed:10882114, PubMed:12234937, PubMed:27720643, PubMed:32494006). The 17S U2 SnRNP complex (1) directly participates in early spliceosome assembly and (2) mediates recognition of the intron branch site during pre-mRNA splicing by promoting the selection of the pre-mRNA branch-site adenosine, the nucleophile for the first step of splicing (PubMed:12234937
Nucleus
Acrofacial dysostosis 1, Nager type
A form of acrofacial dysostosis, a group of disorders which are characterized by malformation of the craniofacial skeleton and the limbs. The major facial features of AFD1 include downslanted palpebral fissures, midface retrusion, and micrognathia, the latter of which often requires the placement of a tracheostomy in early childhood. Limb defects typically involve the anterior (radial) elements of the upper limbs and manifest as small or absent thumbs, triphalangeal thumbs, radial hyoplasia or aplasia, and radioulnar synostosis. Phocomelia of the upper limbs and, occasionally, lower-limb defects have also been reported.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Plays a critical role in bone formation and skeletal development. May be involved in early embryonic morphogenesis
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membraneNucleus
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Required for pre-mRNA splicing as component of the spliceosome, including pre-catalytic, catalytic and post-catalytic spliceosomal complexes (PubMed:25092792, PubMed:28076346, PubMed:28502770, PubMed:28781166, PubMed:29301961, PubMed:29360106, PubMed:29361316, PubMed:30315277, PubMed:30705154). Component of the U5 snRNP and the U4/U6-U5 tri-snRNP complex, a building block of the spliceosome (PubMed:16723661). As a component of the minor spliceosome, involved in the splicing of U12-type introns i
Nucleus
Mandibulofacial dysostosis with microcephaly
A rare syndrome characterized by progressive microcephaly, midface and malar hypoplasia, micrognathia, microtia, dysplastic ears, preauricular skin tags, significant developmental delay, and speech delay. Many patients have major sequelae, including choanal atresia that results in respiratory difficulties, conductive hearing loss, and cleft palate.
involved in nervous system development, important for striatal morphology and motor regulation
Acromelic frontonasal dysostosis
A rare variant form of frontonasal dysplasia, an array of abnormalities affecting the eyes, forehead and nose and linked to midfacial dysraphia. The clinical picture is highly variable. Major findings include true ocular hypertelorism, broadening of the nasal root, median facial cleft affecting the nose and/or upper lip and palate, unilateral or bilateral clefting of the alae nasi, lack of formation of the nasal tip, anterior cranium bifidum occultum, a V-shaped or widow's peak frontal hairline. AFND is characterized by the association of frontonasal malformations with various combinations of polydactyly, tibial hypoplasia, epibulbar dermoid, encephalocoele, corpus callosum agenesis and Dandy-Walker malformation.
Catalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Required for UMP biosynthesis via de novo pathway
Mitochondrion inner membrane
Postaxial acrofacial dysostosis
An autosomal recessive syndrome characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Involved in endochondral growth and skeletal development
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membrane
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Catalytic core component of RNA polymerase I (Pol I), a DNA-dependent RNA polymerase which synthesizes ribosomal RNA precursors using the four ribonucleoside triphosphates as substrates. Transcribes 47S pre-rRNAs from multicopy rRNA gene clusters, giving rise to 5.8S, 18S and 28S ribosomal RNAs (PubMed:11250903, PubMed:11283244, PubMed:16858408, PubMed:34671025, PubMed:34887565, PubMed:36271492). Pol I-mediated transcription cycle proceeds through transcription initiation, transcription elongati
Nucleus, nucleolusChromosome
Acrofacial dysostosis, Cincinnati type
A form of acrofacial dysostosis, a group of disorders characterized by malformations of the craniofacial skeleton and, in some patients, the limbs. AFDCIN patients may also have structural cardiac defects and neurologic abnormalities including developmental delay, hypotonia, motor delay and seizures. AFDCIN inheritance is autosomal dominant.
Key downstream component of the canonical Wnt signaling pathway (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:18957423, PubMed:21262353, PubMed:22155184, PubMed:22647378, PubMed:22699938). In the absence of Wnt, forms a complex with AXIN1, AXIN2, APC, CSNK1A1 and GSK3B that promotes phosphorylation on N-terminal Ser and Thr residues and ubiquitination of CTNNB1 via BTRC and its subsequent degradation by the proteasome (PubMed:17524503, PubMed:18077326, PubMed:18086858, PubMed:189574
CytoplasmNucleusCytoplasm, cytoskeletonCell junction, adherens junctionCell junctionCell membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindle poleSynapseCytoplasm, cytoskeleton, cilium basal body
Colorectal cancer
A complex disease characterized by malignant lesions arising from the inner wall of the large intestine (the colon) and the rectum. Genetic alterations are often associated with progression from premalignant lesion (adenoma) to invasive adenocarcinoma. Risk factors for cancer of the colon and rectum include colon polyps, long-standing ulcerative colitis, and genetic family history.
Variantes genéticas (ClinVar)
840 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 41 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
42 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disostose acrofacial
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
Miller syndrome (MIM#263750) is a rare autosomal recessive acrofacial dysostosis associated with biallelic DHODH variants. Since the identification of the gene in 2010, five case reports have described a variable phenotype in only nine individuals from eight families. We present a cohort of 10 individuals from seven families affected by Miller syndrome, spanning the prenatal stage to 46 years of age. We report on the largest cohort of Miller syndrome to date, which highlights novel findings, including optic atrophy in multiple members of one consanguineous family. The typical postaxial limb defects, including the absence of the 5th digit, were consistent with prior descriptions, but we highlighted the frequent involvement of preaxial structures (thumb and tibial hypoplasia). A higher incidence of camptodactyly and the presence of facial nevus in two patients were notable findings. Congenital heart defects, primarily atrial septal defects, were common, and all living individuals had normal neurodevelopment. This cohort expands the phenotypic spectrum of Miller syndrome associated with variation in DHODH, presenting new findings such as preaxial involvement and facial nevus simplex. Optic atrophy in one family could prompt screening of other cases. Early prenatal diagnosis, particularly in the presence of cardinal limb and cardiac malformations, is crucial for management and genetic counseling.
Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
Nager syndrome (NS) is a rare congenital disorder primarily characterized by mandibulofacial dysostosis and upper limb anomalies. Pathogenic variants in SF3B4, which encodes a core spliceosomal component, represent the primary known genetic cause of NS. This review synthesizes recent findings from cellular, zebrafish, Xenopus, and mouse models to elucidate how SF3B4 deficiency perturbs neural crest cell (NCC) biology and multi-tissue development. Loss of SF3B4 induces widespread splicing abnormalities, with preferential exon skipping affecting AT-rich and GC-poor exons, thereby altering the expression of genes critical for NCC survival, proliferation, migration, and lineage specification. These cellular defects are further exacerbated by oxidative stress and activation of the p53 pathway, resulting in a broad spectrum of developmental abnormalities involving craniofacial, cardiac, skeletal, and sensory (auditory and ocular) systems. Together, these findings highlight the essential role of SF3B4 in coordinating early morphogenesis. Cross-species comparisons reveal conserved NCC vulnerabilities alongside model-specific phenotypes, highlighting the challenge of linking individual splicing alterations to distinct structural outcomes in NS. Future research directions include defining tissue-specific SF3B4-dependent splicing targets, developing human induced pluripotent stem cell-derived models, and exploring therapeutic strategies aimed at restoring splicing homeostasis or compensating for disrupted developmental signaling pathways. This article is categorized under: Congenital Diseases > Molecular and Cellular Physiology Congenital Diseases > Genetics/Genomics/Epigenetics Congenital Diseases > Stem Cells and Development.
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
Management and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
To compare management and outcomes of infants with mandibulofacial dysostosis syndromes (Treacher Collins and Nager syndromes) admitted to neonatal intensive care units (NICUs) to infants with other causes of micrognathia. The Children's Hospitals Neonatal Database from 2010 to 2023 was queried for infants with diagnoses of Treacher Collins syndrome (TCS), Nager syndrome (NS), and other infants in NICUs with micrognathia (n = 4210). We identified 103 infants with TCS and 11 with NS to compare with the micrognathia cohort (n = 4210). Compared with infants with micrognathia, those with TCS were more likely to undergo tracheostomy (54% vs 11%) and gastrostomy tube placement (67% vs 35%) and were less likely to undergo mandibular distraction (9.7% vs 28.2%). The hospital mortality rate in TCS was lower than micrognathia cohort (1.9% vs 7.2%). Apgar scores were similar for TCS and micrognathia cohorts (6 and 8 vs 7 and 8, at 1 and 5 minutes, respectively) but lower for NS (2 and 6). Infants with NS had the highest rate of intubation at birth (91%) and tracheostomy placement (72.7%), and a higher mortality rate than TCS (27.3% vs 1.9%). Hospital length of stay was longer in TCS (47.5 days) and NS (43 days) than the micrognathia cohort (37 days). Infants with mandibulofacial dysostosis (TCS and NS) were more likely to have a tracheostomy and gastrostomy tube, and less likely to undergo mandibular distraction than infants with micrognathia from other causes. NS was most severe with highest mortality rate and lowest Apgar scores. Despite a higher rate of tracheostomy and longer length of stay, the mortality rate for TCS remained low.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
Nager acrofacial dysostosis (#154400) is a rare and mostly sporadic malformation syndrome characterized by craniofacial and extremity findings. In the study, a new case diagnosed in the neonatal period will be presented. The neonatal intensive care unit consulted with our pediatrics genetic clinic for a 2-week-old male patient, who was being followed up in their unit, due to his dysmorphic findings and extremity defects. In the physical examination, downward palpebral fissures, zygomatic bone hypoplasia, mandibular hypoplasia, retromicrognathia, bilateral microtia, bilateral external auditory canal atresia, and bilateral thumb agenesis were observed. Direct radiographs showed left radius hypoplasia and bilateral thumb agenesis. Nager syndrome was considered in the presence of typical craniofacial findings, preaxial extremity deformities, and radiological findings. In the SF3B4 sequence analysis, c.737dupC p.(V247Sfs*239) heterozygous variant was detected. As a result of the segregation analysis, it was demonstrated that the variant was de novo. Nager acrofacial dysostosis should be considered in the patients with craniofacial malformations and radial ray findings.
Publicações recentes
A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
📚 EuropePMC123 artigos no totalmostrando 74
Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
WIREs mechanisms of diseaseA novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
BMC medical genomicsA New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
Molecular syndromologyThe Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
Clinical geneticsRNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
GenesPTBP3 Associated With 9q32 Locus Is a Candidate Gene for Nager Syndrome.
Birth defects researchFacial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
BiologyThe Development of a European Registry for Facial Dysostosis Syndromes: A Delphi-Guided Approach.
The Journal of craniofacial surgeryUnveiling the Phenotypic Spectrum of Miller Syndrome: A Systematic Review.
The Journal of craniofacial surgeryManagement and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
The Journal of pediatricsHuman stem cell model of neural crest cell differentiation reveals a requirement of SF3B4 in survival, maintenance, and differentiation.
Developmental dynamics : an official publication of the American Association of AnatomistsA mild skeletal phenotype with overlapping features of Miller syndrome and functional characterisation of two new variants of human dihydroorotate dehydrogenase.
HeliyonSevere Phenotype With RECQL4 Syndrome: A Report of Two Cases.
American journal of medical genetics. Part AVariant characterisation and clinical profile in a large cohort of patients with Ellis-van Creveld syndrome and a family with Weyers acrofacial dysostosis.
Journal of medical geneticsTranscriptomic analysis reveals mitochondrial dysfunction in the pathogenesis of Nager syndrome in sf3b4-depleted zebrafish.
Biochimica et biophysica acta. Molecular basis of diseaseWeyers Acrofacial Dysostosis: A Case Report.
CureusChildren with Rare Nager Syndrome-Literature Review, Clinical and Physiotherapeutic Management.
GenesEVC-EVC2 complex stability and ciliary targeting are regulated by modification with ubiquitin and SUMO.
Frontiers in cell and developmental biologyAssessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population.
Molecular genetics & genomic medicinePOLR1A variants underlie phenotypic heterogeneity in craniofacial, neural, and cardiac anomalies.
American journal of human geneticsFirst case report of Nager syndrome patient from Georgia.
SAGE open medical case reportsDynamic regulation and requirement for ribosomal RNA transcription during mammalian development.
Proceedings of the National Academy of Sciences of the United States of AmericaMicrodeletion of 4p16.2 in Children: A Case Report and Literature Review.
Case reports in geneticsSF3B4 Frameshift Variants Represented a More Severe Clinical Manifestation in Nager Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationPhenotypic and Molecular Heterogeneity in Mandibulofacial Dysostoses: A Case Series From India.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationCongenital limb deficiency: Genetic investigation of 44 individuals presenting mainly longitudinal defects in isolated or syndromic forms.
Clinical geneticsMolecular mechanisms of hearing loss in Nager syndrome.
Developmental biologyThe role of double-step advancement genioplasty and bilateral coronoidectomy in Nager Syndrome: A case report.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryNager syndrome in patient lacking acrofacial dysostosis: Expanding the phenotypic spectrum of SF3B4-related disease.
American journal of medical genetics. Part ATargeted Next-Generation Sequencing in the Diagnosis of Facial Dysostoses.
Frontiers in geneticsMandibulofacial dysostosis with microcephaly: An expansion of the phenotype via parental survey.
American journal of medical genetics. Part ABroad-spectrum next-generation sequencing-based diagnosis of a case of Nager syndrome.
Journal of clinical laboratory analysisHyoid Bone Position and Head Posture in Patients With Richieri-Costa Pereira Syndrome (EIF4A3 Mutations).
The Journal of craniofacial surgeryAssociated syndromes in patients with Pierre Robin Sequence.
International journal of pediatric otorhinolaryngologyHeterozygous mutation of the splicing factor Sf3b4 affects development of the axial skeleton and forebrain in mouse.
Developmental dynamics : an official publication of the American Association of AnatomistsErosive pustular dermatosis of the scalp in an adolescent with near-total hair regrowth: Case report and review of the literature.
Pediatric dermatologyA Case of Nager Syndrome Diagnosed Before Birth.
Acta medica OkayamaGenetic Polymorphisms Associated with Idiopathic Short Stature and First-Year Response to Growth Hormone Treatment.
Hormone research in paediatricsThe final demise of Rodriguez lethal acrofacial dysostosis: A case report and review of the literature.
American journal of medical genetics. Part AComplexity of the 5' Untranslated Region of EIF4A3, a Critical Factor for Craniofacial and Neural Development.
Frontiers in geneticsModified Lefort Distraction Osteogenesis for the Treatment of Nager Syndrome-Associated Midface Hypoplasia: Technique and Review.
The Journal of craniofacial surgeryCephalometric Findings in Nine Individuals With Richieri-Costa-Pereira Syndrome.
The Journal of craniofacial surgeryNovel mutation in EFCAB7 alters expression and interaction of Ellis-van Creveld ciliary proteins.
Congenital anomaliestp53-dependent and independent signaling underlies the pathogenesis and possible prevention of Acrofacial Dysostosis-Cincinnati type.
Human molecular geneticsDecannulation and Airway Outcomes With Maxillomandibular Distraction in Treacher Collins and Nager Syndrome.
The Journal of craniofacial surgeryRichieri-Costa-Pereira syndrome: Expanding its phenotypic and genotypic spectrum.
Clinical geneticsAnkylosis of temporomandibular joints after mandibular distraction osteogenesis in patients with Nager syndrome: Report of two cases and literature review.
Journal of plastic, reconstructive & aesthetic surgery : JPRASRole of Primary Cilia in Odontogenesis.
Journal of dental researchRare syndromes of the head and face: mandibulofacial and acrofacial dysostoses.
Wiley interdisciplinary reviews. Developmental biologySynchronous Bilateral Breast Cancer in a Patient With Nager Syndrome.
Clinical breast cancerA synonymous splicing mutation in the SF3B4 gene segregates in a family with highly variable Nager syndrome.
European journal of human genetics : EJHGMitochondrial nucleic acid binding proteins associated with diseases.
Frontiers in bioscience (Landmark edition)[Analysis of causes and whole microbial structure in a case of rampant caries].
Nan fang yi ke da xue xue bao = Journal of Southern Medical UniversityDoes canal wall down mastoidectomy benefit syndromic children with congenital aural stenosis?
International journal of pediatric otorhinolaryngologyA Case Report of Absent Epiglottis in Children With Nager Syndrome: Its Impact on Swallowing.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationRodriguez acrofacial dysostosis is caused by apparently de novo heterozygous mutations in the SF3B4 gene.
American journal of medical genetics. Part AAltered mRNA Splicing, Chondrocyte Gene Expression and Abnormal Skeletal Development due to SF3B4 Mutations in Rodriguez Acrofacial Dysostosis.
PLoS geneticsUse of the C-MAC® adult D blade in paediatric patients with Nager syndrome.
Anaesthesia and intensive careRodriguez lethal acrofacial dysostosis syndrome with ambiguous genitalia.
Taiwanese journal of obstetrics & gynecologyElevated plasma dihydroorotate in Miller syndrome: Biochemical, diagnostic and clinical implications, and treatment with uridine.
Molecular genetics and metabolismThe Craniofacial and Upper Limb Management of Nager Syndrome.
The Journal of craniofacial surgeryPropranolol-induced gingival hyperplasia with Nager syndrome: A rare adverse drug reaction.
Journal of advanced pharmaceutical technology & researchNager Syndrome with Eventration of Diaphragm: A Rare Presentation.
Indian journal of pediatricsSf3b4-depleted Xenopus embryos: A model to study the pathogenesis of craniofacial defects in Nager syndrome.
Developmental biologyTruncation and microdeletion of EVC/EVC2 with missense mutation of EFCAB7 in Ellis-van Creveld syndrome.
Congenital anomaliesNager acrofacial dysostosis: a rare genetic disorder causing bilateral temperomandibular joint ankylosis in a 10-year-old girl.
BMJ case reportsPrenatal diagnosis of Nager syndrome in a 12-week-old fetus with a whole gene deletion of SF3B4 by chromosomal microarray.
European journal of medical geneticsNovel mutations in EVC cause aberrant splicing in Ellis-van Creveld syndrome.
Molecular genetics and genomics : MGGDental Management of a Patient with Nager Acrofacial Dysostosis.
Case reports in dentistry[Nager syndrome associated with tetralogy of Fallot: A frequent association?].
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieAcrofacial Dysostosis, Cincinnati Type, a Mandibulofacial Dysostosis Syndrome with Limb Anomalies, Is Caused by POLR1A Dysfunction.
American journal of human geneticsA review of craniofacial disorders caused by spliceosomal defects.
Clinical geneticsNager syndrome and Pierre Robin sequence.
Pediatrics international : official journal of the Japan Pediatric SocietyOrbital soft tissue surgery for patients with Treacher-Collins or Nager syndrome. A new surgical approach with early correction of soft tissue: prospective study.
The British journal of oral & maxillofacial surgeryAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disostose acrofacial.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disostose acrofacial
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- The Phenotypic Spectrum of Miller Syndrome: Insight From a French Cohort.
- Nager Syndrome Revisited: Integrating In Vivo and In Vitro Models to Decipher SF3B4-Dependent Tissue Coordination.
- A novel EVC2 splice-site variant expands the mutational and phenotypic spectrum of Weyers acrofacial dysostosis.
- Management and Outcomes of Neonates with Treacher Collins and Nager Syndromes.
- A New Case of Nager Syndrome as a Rare Cause of Acrofacial Dysostosis.
- RNA Polymerase I Dysfunction Underlying Craniofacial Syndromes: Integrated Genetic Analysis Reveals Parallels to 22q11.2 Deletion Syndrome.
- Facial Bone Defects Associated with Lateral Facial Clefts Tessier Type 6, 7 and 8 in Syndromic Neurocristopathies: A Detailed Micro-CT Analysis on Historical Museum Specimens.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:364574(Orphanet)
- MONDO:0018237(MONDO)
- GARD:21574(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21082523(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Disostose acrofacial
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov