A Disostose Acrofacial Pós-Axial (POADS) é uma condição que causa malformações nos ossos do rosto e das extremidades (braços e pernas). Ela é caracterizada por: queixo e maçãs do rosto pouco desenvolvidos; orelhas pequenas e em forma de concha; a pálpebra inferior virada para fora; e deficiências simétricas nos braços e pernas que afetam o lado do dedo mínimo. Essas deficiências incluem a ausência do quinto raio digital (o dedo mínimo e os ossos correspondentes da mão/pé) e o desenvolvimento incompleto da ulna (o osso do antebraço localizado no lado do dedo mínimo).
Introdução
O que você precisa saber de cara
A Disostose Acrofacial Pós-Axial (POADS) é uma condição que causa malformações nos ossos do rosto e das extremidades (braços e pernas). Ela é caracterizada por: queixo e maçãs do rosto pouco desenvolvidos; orelhas pequenas e em forma de concha; a pálpebra inferior virada para fora; e deficiências simétricas nos braços e pernas que afetam o lado do dedo mínimo. Essas deficiências incluem a ausência do quinto raio digital (o dedo mínimo e os ossos correspondentes da mão/pé) e o desenvolvimento incompleto da ulna (o osso do antebraço localizado no lado do dedo mínimo).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 40 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalyzes the conversion of dihydroorotate to orotate with quinone as electron acceptor. Required for UMP biosynthesis via de novo pathway
Mitochondrion inner membrane
Postaxial acrofacial dysostosis
An autosomal recessive syndrome characterized by severe micrognathia, cleft lip and/or palate, hypoplasia or aplasia of the posterior elements of the limbs, coloboma of the eyelids and supernumerary nipples.
Variantes genéticas (ClinVar)
53 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Disostose acrofacial pós-axial
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
A mild skeletal phenotype with overlapping features of Miller syndrome and functional characterisation of two new variants of human dihydroorotate dehydrogenase.
Dihydroorotate dehydrogenase (DHODH) catalyzes the fourth enzymatic reaction of the pyrimidine biosynthesis pathway. Miller syndrome, also known as postaxial acrofacial dysostosis, is caused by biallelic pathogenic variants in DHODH. We present a patient with a relatively mild skeletal phenotype carrying a novel variant of unknown significance in DHODH: c.829G > A, p.(D277N), in combination with a known variant, c.403C > T, p.(R135C). We functionally characterized the DHODH variant D277N in comparison to a very recently reported, but functionally uncharacterized variant P43L, that was found in a patient with more pronounced Miller syndrome features. Because both cases share the same DHODH variant R135C, we aimed to study the effect on enzyme activity of the two variants D277N and P43L to determine pathogenicity and possibly a genotype-phenotype relationship on the R135C background. We found a significant reduction in enzyme activity for both variants. The variant P43L showed a more pronounced loss of function in all assays compatible with other pathogenic variants reported in Miller, whereas the D277N variant showed milder changes that could reflect the mild phenotypic features in our patient. Yet due to a lack of a known threshold of residual enzyme activity to determine pathogenicity, this needs to be confirmed in further studies.
Assessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population.
Miller syndrome is a rare type of postaxial acrofacial dysostosis caused by biallelic mutations in the DHODH gene, which is characterized mainly by craniofacial malformations of micrognathia, orofacial clefts, cup-shaped ears, and malar hypoplasia, combined with postaxial limb deformities like the absence of fifth digits. In this study, a prenatal case with multiple orofacial-limb abnormities was enrolled, and a thorough clinical and imaging examination was performed. Subsequently, genetic detection with karyotyping, chromosomal microarray analysis (CMA) and whole-exome sequencing (WES) was carried out. In vitro splicing analysis was also conducted to clarify the impact of one novel variant. The affected fetus displayed typical manifestations of Miller syndrome, and WES identified a diagnostic compound heterozygous variation in DHODH, consisting of two variants: exon(1-3)del and c.819 + 5G > A. We conducted a further in vitro validation with minigene system, and the result indicated that the c.819 + 5G > A variant would lead to an exon skipping in mRNA splicing. These findings provided with the first exonic deletion and first splice site variant in DHODH, which expanded the mutation spectrum of Miller syndrome and offered reliable evidence for genetic counseling to the affected family.
Mitochondrial nucleic acid binding proteins associated with diseases.
Mammalian mitochondrial DNA (mtDNA) exists in structures called nucleoids, which correspond to the configuration of nuclear DNA. Mitochondrial transcription factor A (TFAM), first cloned as an mtDNA transcription factor, is critical for packaging and maintaining mtDNA. To investigate functional aspects of TFAM, we identified many RNA-binding proteins as candidate TFAM interactors, including ERAL1 and p32. In this review, we first describe the functions of TFAM, replication proteins such as polymerase gamma and Twinkle, and mitochondrial RNA binding proteins. We describe the role of mitochondrial nucleic acid binding proteins within the mitochondrial matrix and two oxidative phosphorylation-related proteins within the mitochondrial intermembrane space. We then discuss how mitochondrial dysfunction is related to several diseases, including mitochondrial respiratory disease, Miller syndrome and cancer. We also describe p32 knockout mice, which are embryonic lethal and exhibit respiratory chain defects. Miller syndrome is a recessive disorder characterized by postaxial acrofacial dysostosis and caused by a mutation in DHODH. Finally, we explain that p32 and mitochondrial creatine kinase may be novel markers for the progression of prostate cancer.
Publicações recentes
A mild skeletal phenotype with overlapping features of Miller syndrome and functional characterisation of two new variants of human dihydroorotate dehydrogenase.
Assessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population.
Mitochondrial nucleic acid binding proteins associated with diseases.
Orodental findings in postaxial acrofacial dysostosis.
Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction.
📚 EuropePMC23 artigos no totalmostrando 3
A mild skeletal phenotype with overlapping features of Miller syndrome and functional characterisation of two new variants of human dihydroorotate dehydrogenase.
HeliyonAssessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population.
Molecular genetics & genomic medicineMitochondrial nucleic acid binding proteins associated with diseases.
Frontiers in bioscience (Landmark edition)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Disostose acrofacial pós-axial.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Disostose acrofacial pós-axial
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A mild skeletal phenotype with overlapping features of Miller syndrome and functional characterisation of two new variants of human dihydroorotate dehydrogenase.
- Assessment of a novel variation in DHODH gene causing Miller syndrome: The first report in Chinese population.
- Mitochondrial nucleic acid binding proteins associated with diseases.
- Orodental findings in postaxial acrofacial dysostosis.
- Dihydro-orotate dehydrogenase is physically associated with the respiratory complex and its loss leads to mitochondrial dysfunction.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:246(Orphanet)
- OMIM OMIM:263750(OMIM)
- MONDO:0009903(MONDO)
- GARD:8410(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q2691713(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
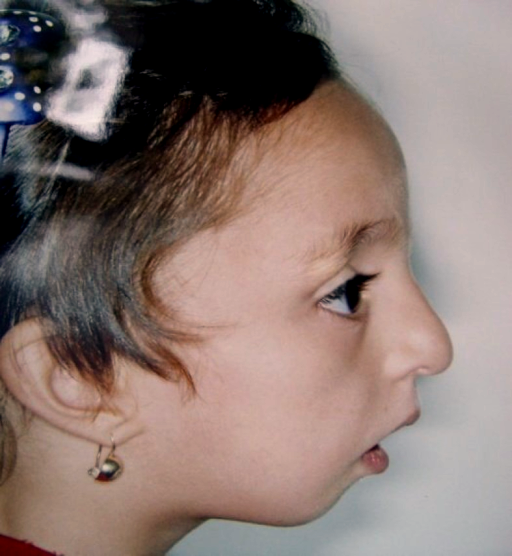
Disostose acrofacial pós-axial
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov