Síndrome de Kagami-Ogata, causada por dissomia uniparental paterna do cromossomo 14. Caracteriza-se por polegar aduzido, hipotonia axial, dedos longos e anomalias cardíacas como estenose pulmonar e persistência do canal arterial.
Introdução
O que você precisa saber de cara
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 14, também conhecida como síndrome de Kagami-Ogata, é uma doença genética rara caracterizada por um conjunto de sinais e sintomas que afetam múltiplos sistemas do corpo. A condição é causada por uma alteração cromossômica específica e tem prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. O início das manifestações ocorre no período neonatal ou na infância.[1][4]
Sinais e sintomas
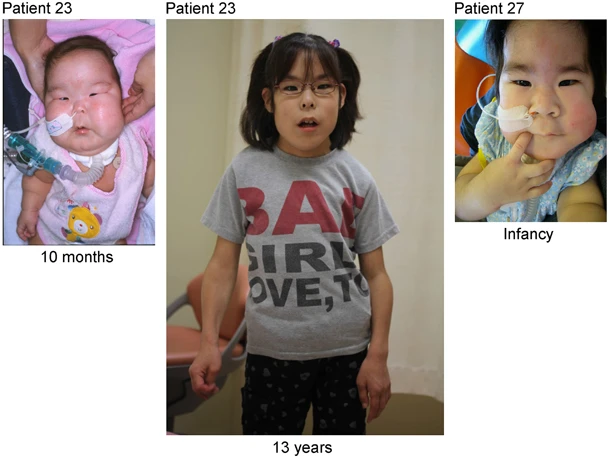
Os sinais e sintomas da síndrome de Kagami-Ogata são variados e podem incluir: hipotonia (diminuição do tônus muscular) generalizada e axial, atraso global do desenvolvimento, polegares aduzidos (voltados para dentro), dedos longos, metatarso aduto (pés virados para dentro), tórax em sino (formato de sino), pectus excavatum (tórax escavado), anormalidades da junção costocondral, costelas finas e ossos wormianos (ossos extras no crânio). Também podem ocorrer alterações cardíacas como estenose pulmonar, persistência do canal arterial, defeito do septo atrial e hipertensão arterial pulmonar. Outros achados incluem abdome protuberante, diástase dos retos (separação dos músculos abdominais), hérnia inguinal, esplenomegalia (aumento do baço), distância intermamilar ampla, hipoplasia genital externa, microcefalia, retardo do crescimento pós-natal e polidrâmnio (excesso de líquido amniótico durante a gestação).[1][4]
Causas genéticas
A doença é causada pela dissomia uniparental paterna do cromossomo 14, ou seja, a pessoa herda duas cópias do cromossomo 14 do pai e nenhuma da mãe. Essa alteração genética afeta a expressão de genes localizados em uma região específica do cromossomo 14 (14q32), que sofrem imprinting genômico. Os genes envolvidos incluem MEG3, DLK1 (que codifica a proteína delta homolog 1) e RTL1 (que codifica a proteína retrotransposon-like protein 1). A herança é considerada autossômica dominante, mas na prática a condição geralmente ocorre de forma esporádica (não herdada).[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais e sintomas e confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína pode ser utilizada como exame complementar. No Brasil, esses procedimentos fazem parte da cobertura mínima do SUS para doenças raras. Atualmente, há 129 variantes associadas à doença registradas no ClinVar e 10 testes genéticos disponíveis.[1][2][5]
Tratamento e manejo
O tratamento é multidisciplinar e focado no manejo dos sintomas específicos de cada paciente. Não há medicamentos específicos aprovados para a síndrome de Kagami-Ogata. O acompanhamento deve incluir atendimento em reabilitação para doenças raras, conforme disponível no SUS. O suporte pode envolver fisioterapia, terapia ocupacional, fonoaudiologia e acompanhamento com cardiologista, geneticista e outros especialistas conforme a necessidade.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade dos sintomas, especialmente os cardíacos e respiratórios. O acompanhamento médico regular e o suporte multidisciplinar são fundamentais para melhorar a qualidade de vida e o desenvolvimento da pessoa com a síndrome. O atraso global do desenvolvimento e a hipotonia podem exigir intervenções precoces e contínuas.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Síndrome de Kagami-Ogata, causada por dissomia uniparental paterna do cromossomo 14. Caracteriza-se por polegar aduzido, hipotonia axial, dedos longos e anomalias cardíacas como estenose pulmonar e persistência do canal arterial.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Dissomia uniparental de origem paterna, cromossomo 14, também conhecida como síndrome de Kagami-Ogata, é uma doença genética rara caracterizada por um conjunto de sinais e sintomas que afetam múltiplos sistemas do corpo. A condição é causada por uma alteração cromossômica específica e tem prevalência estimada em menos de 1 caso por 1.000.000 de pessoas. O início das manifestações ocorre no período neonatal ou na infância.[1][4]
Sinais e sintomas
Os sinais e sintomas da síndrome de Kagami-Ogata são variados e podem incluir: hipotonia (diminuição do tônus muscular) generalizada e axial, atraso global do desenvolvimento, polegares aduzidos (voltados para dentro), dedos longos, metatarso aduto (pés virados para dentro), tórax em sino (formato de sino), pectus excavatum (tórax escavado), anormalidades da junção costocondral, costelas finas e ossos wormianos (ossos extras no crânio). Também podem ocorrer alterações cardíacas como estenose pulmonar, persistência do canal arterial, defeito do septo atrial e hipertensão arterial pulmonar. Outros achados incluem abdome protuberante, diástase dos retos (separação dos músculos abdominais), hérnia inguinal, esplenomegalia (aumento do baço), distância intermamilar ampla, hipoplasia genital externa, microcefalia, retardo do crescimento pós-natal e polidrâmnio (excesso de líquido amniótico durante a gestação).[1][4]
Causas genéticas
A doença é causada pela dissomia uniparental paterna do cromossomo 14, ou seja, a pessoa herda duas cópias do cromossomo 14 do pai e nenhuma da mãe. Essa alteração genética afeta a expressão de genes localizados em uma região específica do cromossomo 14 (14q32), que sofrem imprinting genômico. Os genes envolvidos incluem MEG3, DLK1 (que codifica a proteína delta homolog 1) e RTL1 (que codifica a proteína retrotransposon-like protein 1). A herança é considerada autossômica dominante, mas na prática a condição geralmente ocorre de forma esporádica (não herdada).[1][2][5]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sinais e sintomas e confirmado por testes genéticos. Os exames disponíveis incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; e sequenciamento completo do exoma (WES). A dosagem de alfa-fetoproteína pode ser utilizada como exame complementar. No Brasil, esses procedimentos fazem parte da cobertura mínima do SUS para doenças raras. Atualmente, há 129 variantes associadas à doença registradas no ClinVar e 10 testes genéticos disponíveis.[1][2][5]
Tratamento e manejo
O tratamento é multidisciplinar e focado no manejo dos sintomas específicos de cada paciente. Não há medicamentos específicos aprovados para a síndrome de Kagami-Ogata. O acompanhamento deve incluir atendimento em reabilitação para doenças raras, conforme disponível no SUS. O suporte pode envolver fisioterapia, terapia ocupacional, fonoaudiologia e acompanhamento com cardiologista, geneticista e outros especialistas conforme a necessidade.[1][2]
Prognóstico e qualidade de vida
O prognóstico varia de acordo com a gravidade dos sintomas, especialmente os cardíacos e respiratórios. O acompanhamento médico regular e o suporte multidisciplinar são fundamentais para melhorar a qualidade de vida e o desenvolvimento da pessoa com a síndrome. O atraso global do desenvolvimento e a hipotonia podem exigir intervenções precoces e contínuas.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 53 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 130 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição.
May have a role in neuroendocrine differentiation
MembraneCytoplasm
Plays an essential role in capillaries endothelial cells for the maintenance of feto-maternal interface and for development of the placenta
Membrane
Variantes genéticas (ClinVar)
129 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Dissomia uniparental de origem paterna, cromossomo 14
Centros de Referência SUS
24 centros habilitados pelo SUS para Dissomia uniparental de origem paterna, cromossomo 14
Centros para Dissomia uniparental de origem paterna, cromossomo 14
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
Kagami-Ogata syndrome (KOS) is a clinically recognizable syndrome in the neonatal period. It is characterized by specific skeletal anomalies and facial dysmorphisms. It is typically caused by paternal uniparental disomy of chromosome 14, while epimutations and microdeletions are less commonly reported causes. In the pediatric setting, KOS is a well delineated syndrome. However, there is a dearth of literature describing the natural history of the condition in adults. Herein, we describe a 35-year-old man, the first adult with KOS reported due to paternal uniparental disomy 14, and review reports of KOS in other affected adults. This highlights the variability in neurocognitive phenotypes, the presence of connective tissue abnormalities, and the uncertainties around long-term cancer risk.
Maternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype.
Kagami-Ogata syndrome and Temple syndrome are imprinting disorders caused by the abnormal expression of genes in an imprinted cluster on chromosome 14q32. Here, we report a female with mild features of the Kagami-Ogata syndrome phenotype with polyhydramnios, neonatal hypotonia, feeding difficulties, abnormal foot morphology, patent foramen ovale, distal arthrogryposis, normal facial profile, and a bell-shaped thorax without coat hanger ribs. The single nucleotide polymorphism array revealed the interstitial deletion of chromosome 14q32.2-q32.31 (117 kb in size), involving the RTL1as and MEG8 genes, and other small nucleolar RNAs and microRNAs. The differentially methylated regions (DMRs) appeared unaltered. The RTL1as gene deletion and the normal methylation pattern of the MEG3 gene loci were confirmed by methylation-specific multiplex ligation-dependent probe amplification. Deletions of the 14q32 region without involving DMRs, and encompassing only the RTL1as and MEG8 genes, are poorly described in the literature. The mother's chromosomal microarray also confirmed the identical 14q32.2 deletion, although she presented a normal phenotype. The maternally inherited 14q32 deletion was responsible for Kagami-Ogata syndrome in our patient. It was not sufficient, however, to produce Temple syndrome or any other pathogenic phenotype in the patient's mother.
Paternal UPD14 with sSMC derived from chromosome 14 in Kagami-Ogata syndrome.
Quantitative assessment of coat-hanger ribs detected on three-dimensional ultrasound for prenatal diagnosis of Kagami-Ogata syndrome.
Kagami-Ogata syndrome (KOS14) is a rare disease characterized by omphalocele, polyhydramnios and a bell-shaped thorax. Although the coat-hanger appearance of the ribs on postnatal X-rays is a key diagnostic finding of KOS14, its prenatal diagnosis remains challenging. We encountered a case of KOS14 diagnosed prenatally that showed omphalocele, polyhydramnios, and a bell-shaped narrow thorax. The coat-hanger angle (CHA) measured at the sixth thoracic vertebrae and the ribs using three-dimensional (3D) ultrasonography was 39°, reflecting the coat-hanger appearance of the ribs. Segmental uniparental disomy chromosome 14 (UPD(14)pat) was confirmed by a methylation analysis and microsatellite analysis after birth. The median CHA (minimum, maximum) in 25 normal fetuses was 19 (9, 26) degrees, and a sonographic CHA of 30° may be a border value for diagnosing KOS14. When the combination of omphalocele and polyhydramnios is found prenatally, 3D ultrasonography for CHA might aid in the differential diagnosis of KOS14.
Two infants with mild, atypical clinical features of Kagami-Ogata syndrome caused by epimutation.
Kagami-Ogata syndrome (KOS) is an imprinting disorder characterized by polyhydramnios, bell-shaped thorax with coat-hanger appearance (curved ribs), respiratory distress, abdominal wall defects, and distinct facial features, together with intellectual developmental delay with special needs. Abnormal expression of the imprinted genes on chromosome 14q32.2 causes KOS. Epimutation with aberrant hypermethylation of the MEG3/DLK1: intergenic differentially methylated region (MEG3/DLK1:IG-DMR) and the MEG3:TSS-DMR is one of the etiologies of KOS. We report two infants with KOS caused by epimutation presenting with some characteristic clinical features, mild clinical course, and almost normal motor and intellectual development. Methylation analysis for ten DMRs related to major imprinting disorders using pyrosequencing with genomic DNA (gDNA) extracted from leukocytes showed abnormally increased methylation levels of the MEG3/DLK1:IG-DMR and MEG3:TSS-DMR in both patients, but lower than those in patients with paternal uniparental disomy chromosome 14 (upd(14)pat). The methylation levels in the DMRs other than both DMRs were within normal range. We also conducted methylation analysis for the MEG3/DLK1:IG-DMR and MEG3:TSS-DMR with gDNA extracted from nails and buccal cells of both patients. Methylation levels in the MEG3:TSS-DMR, particularly in buccal cells, were closer to normal range compared to those in leukocytes. Microsatellite analysis for chromosome 14 and array comparative hybridization analysis showed no upd(14)pat or microdeletion involving the 14q32.2 imprinted region in either patient. A differential mosaic ratio of cells with aberrant methylation of DMRs at the 14q32.2 imprinted region among tissues (connective tissue, lung, and brain) might have led to their atypical clinical features. Further studies of patients with epimutation should further expand the phenotypic spectrum of KOS.
Publicações recentes
Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
Maternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype.
Paternal UPD14 with sSMC derived from chromosome 14 in Kagami-Ogata syndrome.
Quantitative assessment of coat-hanger ribs detected on three-dimensional ultrasound for prenatal diagnosis of Kagami-Ogata syndrome.
Two infants with mild, atypical clinical features of Kagami-Ogata syndrome caused by epimutation.
📚 EuropePMCmostrando 21
Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
American journal of medical genetics. Part AMaternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype.
American journal of medical genetics. Part APaternal UPD14 with sSMC derived from chromosome 14 in Kagami-Ogata syndrome.
Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biologyQuantitative assessment of coat-hanger ribs detected on three-dimensional ultrasound for prenatal diagnosis of Kagami-Ogata syndrome.
The journal of obstetrics and gynaecology researchTwo infants with mild, atypical clinical features of Kagami-Ogata syndrome caused by epimutation.
European journal of medical geneticsKagami-Ogata syndrome: a case report.
Journal of medical case reportsPreimplantation genetic testing for a chr14q32 microdeletion in a family with Kagami-Ogata syndrome and Temple syndrome.
Journal of medical geneticsPrenatal diagnosis of Kagami-Ogata syndrome.
Journal of clinical ultrasound : JCUKagami-Ogata syndrome in a patient with 46,XX,t(2;14)(q11.2;q32.2)mat disrupting MEG3.
Journal of human geneticsKagami-Ogata syndrome: an important differential diagnosis to Beckwith-Wiedemann syndrome.
Journal of clinical ultrasound : JCU[Kagami-Ogata Syndrome: An Anomaly of the Ribs as a Pathognomonic Feature for the Clinical Diagnosis of an (epi)Genetic Syndrome].
Zeitschrift fur Geburtshilfe und NeonatologiePrenatal diagnosis of paternal uniparental disomy for chromosome 14 using a single-nucleotide-polymorphism-based microarray analysis: A case report.
Journal of the Formosan Medical Association = Taiwan yi zhiUniparental disomy and prenatal phenotype: Two case reports and review.
MedicineDLK1-DIO3 imprinted locus deregulation in development, respiratory disease, and cancer.
Expert review of respiratory medicineGenome-wide multilocus imprinting disturbance analysis in Temple syndrome and Kagami-Ogata syndrome.
Genetics in medicine : official journal of the American College of Medical GeneticsNovel microdeletions on chromosome 14q32.2 suggest a potential role for non-coding RNAs in Kagami-Ogata syndrome.
European journal of human genetics : EJHGGenomic imprinting of DIO3, a candidate gene for the syndrome associated with human uniparental disomy of chromosome 14.
European journal of human genetics : EJHGPaternal Uniparental Disomy of Chromosome 14 with Hypospadias.
Cytogenetic and genome researchPaternal uniparental disomy chromosome 14-like syndrome due a maternal de novo 160 kb deletion at the 14q32.2 region not encompassing the IG- and the MEG3-DMRs: Patient report and genotype-phenotype correlation.
American journal of medical genetics. Part AComprehensive clinical studies in 34 patients with molecularly defined UPD(14)pat and related conditions (Kagami-Ogata syndrome).
European journal of human genetics : EJHGPrenatal findings and epimutations for paternal uniparental disomy for chromosome 14 syndrome.
The journal of obstetrics and gynaecology researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Dissomia uniparental de origem paterna, cromossomo 14.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Dissomia uniparental de origem paterna, cromossomo 14
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Adults with paternal UPD14 causing Kagami-Ogata syndrome: Case report and review of the literature.
- Maternally inherited deletion encompassing the RTL1as and MEG8 genes of the human 14q32 imprinted region in a patient with a mild Kagami-Ogata syndrome phenotype.
- Paternal UPD14 with sSMC derived from chromosome 14 in Kagami-Ogata syndrome.Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology· 2023· PMID 36656404mais citado
- Quantitative assessment of coat-hanger ribs detected on three-dimensional ultrasound for prenatal diagnosis of Kagami-Ogata syndrome.
- Two infants with mild, atypical clinical features of Kagami-Ogata syndrome caused by epimutation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:96334(Orphanet)
- OMIM OMIM:608149(OMIM)
- MONDO:0011975(MONDO)
- GARD:5409(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783547(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Dissomia uniparental de origem paterna, cromossomo 14
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata