A Síndrome de Ablefaria-Macrostomia é uma condição de má-formações variadas e extremamente rara, presente desde o nascimento. Ela se caracteriza pela combinação de pálpebras ausentes ou pouco desenvolvidas, boca anormalmente grande, orelhas externas com formato incomum, dedos das mãos e dos pés unidos, características da pele (como pele seca e áspera ou dobras de pele em excesso), cabelos ausentes ou ralos, má-formações nos órgãos genitais e atraso no desenvolvimento (observado em dois terços dos casos). Outros sinais que podem aparecer são: pouco desenvolvimento das maçãs do rosto, mamilos ausentes ou pouco desenvolvidos, alterações no umbigo e crescimento abaixo do normal. Geralmente, a síndrome surge de forma espontânea (não é herdada), embora alguns casos em famílias tenham sido registrados. Ela também apresenta muitas semelhanças com a Síndrome de Fraser.
Introdução
O que você precisa saber de cara
A Síndrome de Ablefaria-Macrostomia é uma condição de má-formações variadas e extremamente rara, presente desde o nascimento. Ela se caracteriza pela combinação de pálpebras ausentes ou pouco desenvolvidas, boca anormalmente grande, orelhas externas com formato incomum, dedos das mãos e dos pés unidos, características da pele (como pele seca e áspera ou dobras de pele em excesso), cabelos ausentes ou ralos, má-formações nos órgãos genitais e atraso no desenvolvimento (observado em dois terços dos casos). Outros sinais que podem aparecer são: pouco desenvolvimento das maçãs do rosto, mamilos ausentes ou pouco desenvolvidos, alterações no umbigo e crescimento abaixo do normal. Geralmente, a síndrome surge de forma espontânea (não é herdada), embora alguns casos em famílias tenham sido registrados. Ela também apresenta muitas semelhanças com a Síndrome de Fraser.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 24 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 64 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisBinds to the E-box consensus sequence 5'-CANNTG-3' as a heterodimer and inhibits transcriptional activation by MYOD1, MYOG, MEF2A and MEF2C. Also represses expression of pro-inflammatory cytokines such as TNFA and IL1B. Involved in postnatal glycogen storage and energy metabolism (By similarity). Inhibits the premature or ectopic differentiation of preosteoblast cells during osteogenesis, possibly by changing the internal signal transduction response of osteoblasts to external growth factors
NucleusCytoplasm
Focal facial dermal dysplasia 3, Setleis type
A form of focal facial dermal dysplasia, a group of developmental defects characterized by bitemporal or preauricular skin lesions resembling aplasia cutis congenita. FFDD3 is characterized by distinctive bitemporal scar-like depressions resembling forceps marks, and additional facial features, including a coarse and leonine appearance, absent eyelashes on both lids or multiple rows on the upper lids, absent Meibomian glands, slanted eyebrows, chin clefting, and hypo- or hyperpigmentation of the skin. Histologically, the bitemporal lesion is an ectodermal dysplasia with near absence of subcutaneous fat, suggesting insufficient migration of neural crest cells into the frontonasal process and the first branchial arch.
Variantes genéticas (ClinVar)
94 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de ablefaro-macrostomia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Infantile epileptic spasms syndrome as a new phenotype in TOP2B deficiency caused by a de novo variant: a case report and literature review.
Background: Type II DNA topoisomerases (EC5.99.1.3) are enzymes that catalyze topological changes during DNA replication and gene transcription in an ATP-dependent manner. Vertebrates have two isoforms: topoisomerase IIα and β. Type II topoisomerase β is encoded by TOP2B. For TOP2B, a number of germline pathogenic variants have been identified as causative for human diseases, including Hoffman syndrome, ablepharon-macrostomia syndrome with immunodeficiency, B-cell immunodeficiency, distal limb anomalies, and urogenital malformations syndrome. To date, only 14 patients with the above diseases from seven families have been reported in PubMed. Herein, we describe an additional case of a child who presented with "infantile epileptic spasms syndrome" (IESS) as the first symptom, B-cell immunodeficiency, dysmorphic facial features, and a pathogenic variant in TOP2B. The c.1901A > G variant in TOP2B is new to our study, which further enriches the genotype of TOP2B deficiency. Our patient manifested as a typical triad: infantile spasms, hypsarrhythmia on electroencephalogram, and developmental arrest at the age of 7 months. Although epilepsy and neurodevelopmental disorders have been reported in patients with TOP2B deficiency, typical IESS has not been described previously. IESS in our patient further expands the phenotype of TOP2B. The patient was started on monthly intravenous immunoglobulin replacement therapy after being diagnosed with TOP2B deficiency and since then has not suffered from severe infections. TOP2B deficiency is a group of heterogeneous diseases, which is ultrarare. The results from our study extend the phenotype and genotype spectrum of TOP2B deficiency. TOP2B may be a causative gene for IESS.
Upper and lower eyelid contour and positional changes after deep skin grafts in ablepharon macrostomia syndrome.
Ablepharon macrostomia syndrome is a rare congenital disorder caused by autosomal-dominant TWIST2 mutations. This condition is characterized by redundant skin, low-set ears, macrostomia, ambiguous genitalia, and underdevelopment of the both upper and lower eyelids. The shortening of the anterior lamella, septum and levator aponeurosis lead to a severe corneal exposure within the first hours of life. Since McCarthy and West's first report in 1977, 21 AMS cases have been documented. We report a new AMS case with a quantitative analysis of palpebral fissure changes following skin grafts over the upper and lower smooth tarsal muscles and lateral tarsorrhaphy.
Modified Reverse Hatchet Flap for Ablepharon-Macrostomia Syndrome.
Ablepharon-macrostomia syndrome is a rare disorder characterized by TWIST2 mutations and anterior lamellar dysgenesis. Timely intervention is critical to prevent exposure keratopathy, corneal ulceration, and permanent vision loss. We report a novel approach to multiplanar eyelid reconstruction in ablepharon-macrostomia syndrome involving use of a modified reverse hatchet flap in 1 lower eyelid along with division at the eyelid margin, recession of the eyelid retractors in conjunction with preputial skin grafting for anterior lamellar restoration in the other 3 eyelids.
Cryptophthalmos: associated syndromes and genetic disorders.
Cryptophthalmos is a rare congenital condition caused by anomalous eyelid development where the eyelid folds do not develop or fail to separate. Cryptophthalmos can be unilateral or bilateral and can occur in isolation or as part of an underlying syndrome. We aim to identify genetic syndromes associated with cryptophthalmos to facilitate genetic diagnosis. We performed a retrospective medical record review of all patients diagnosed with cryptophthalmos followed at a single center between 2000 and 2020. The analysis included medical history, clinical examination findings, and genetic testing results. Thirteen patients were included, 10 (77%) males, mean age of 2.4 years. Eight (61%) had bilateral cryptophthalmos, and 4 (31%) had complete cryptophthalmos. Associated ocular abnormalities included corneal opacities (13/13, 100%), upper eyelid colobomas (12/13, 92%), and microphthalmia/clinical anophthalmia (3/13, 23%). All cases of complete cryptophthalmos had bilateral disease. An underlying clinical or molecular diagnosis was identified in 10/13 (77%) cases, including Fraser syndrome (n = 5), amniotic band syndrome (n = 1), FREM1-related disease (n = 1), Goldenhar versus Schimmelpenning syndrome (n = 1), MOTA syndrome (n = 1), and CELSR2-related disease (n = 1). This is the first report of a possible association between cryptophthalmos and biallelic CELSR2 variants. Children with cryptophthalmos, especially those with extra-ocular involvement, should be referred for comprehensive genetic evaluation.
Ocular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
Ablepharon-macrostomia syndrome (AMS) and Barber-Say syndrome (BSS) are congenital ectodermal dysplasias associated with mutations in the TWIST2 gene. Among the ophthalmic anomalies that occur in these syndromes, underdevelopment of the anterior lamella of the eyelid is a defining feature. Reports of mosaic expression of TWIST2 mutations are extremely rare, with only five confirmed or suspected cases described to date. Mosaic expression of TWIST2 variants is correlated with a less severe phenotype than that reported for the typical expression of TWIST2 variants associated with BSS or AMS. Abnormal development of the anterior lamella appears to be a common feature in all cases of AMS with mosaic expression. Here, we describe the phenotype of a patient with mosaic expression of a TWIST2 mutation that is typically associated with AMS. We additionally describe the surgical approach employed in the treatment of this patient.
Publicações recentes
Infantile epileptic spasms syndrome as a new phenotype in TOP2B deficiency caused by a de novo variant: a case report and literature review.
🥈 ObservacionalUpper and lower eyelid contour and positional changes after deep skin grafts in ablepharon macrostomia syndrome.
🥇 Meta-análiseModified Reverse Hatchet Flap for Ablepharon-Macrostomia Syndrome.
Cryptophthalmos: associated syndromes and genetic disorders.
Ablepharon macrostomia syndrome: Absent prepuce in the first case report in West Africa.
🥈 Observacional📚 EuropePMC25 artigos no totalmostrando 21
Infantile epileptic spasms syndrome as a new phenotype in TOP2B deficiency caused by a de novo variant: a case report and literature review.
Frontiers in pediatricsUpper and lower eyelid contour and positional changes after deep skin grafts in ablepharon macrostomia syndrome.
Orbit (Amsterdam, Netherlands)Modified Reverse Hatchet Flap for Ablepharon-Macrostomia Syndrome.
Ophthalmic plastic and reconstructive surgeryCryptophthalmos: associated syndromes and genetic disorders.
Ophthalmic geneticsAblepharon macrostomia syndrome: Absent prepuce in the first case report in West Africa.
The Nigerian postgraduate medical journalOcular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
Orbit (Amsterdam, Netherlands)A case of ablepharon macrostomia syndrome requiring multidisciplinary care.
Clinical & experimental optometryAn optimized base editor with efficient C-to-T base editing in zebrafish.
BMC biologyAblepharon Macrostomia Syndrome: Rib Cartilage and Fat Grafting for Lower Lid Reconstruction.
The Journal of craniofacial surgeryLaryngo-tracheal stenosis in a woman with ablepharon macrostomia syndrome.
BMC pulmonary medicineLong-Term Results of the Surgical Management of the Upper Eyelids in "Ablepharon"-Macrostomia Syndrome.
Ophthalmic plastic and reconstructive surgeryAblepharon and craniosynostosis in a patient with a localized TWIST1 basic domain substitution.
American journal of medical genetics. Part AProgrammable base editing in zebrafish using a modified CRISPR-Cas9 system.
Methods (San Diego, Calif.)Use of the Masquerade Flap in Ablepharon-Macrostomia Syndrome: A Case Report.
CorneaBarber-Say Syndrome and Ablepharon-Macrostomia Syndrome: A Patient's View.
Molecular syndromologyBarber-say syndrome: a confirmed case of TWIST2 gene mutation.
Clinical case reportsThe focal facial dermal dysplasias: phenotypic spectrum and molecular genetic heterogeneity.
Journal of medical geneticsLocalized TWIST1 and TWIST2 basic domain substitutions cause four distinct human diseases that can be modeled in Caenorhabditis elegans.
Human molecular geneticsBarber-Say syndrome and Ablepharon-Macrostomia syndrome: An overview.
American journal of medical genetics. Part AA Case Report of Ablepharon-Macrostomia Syndrome with Amniotic Membrane Grafting.
Case reports in ophthalmologyRecurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de ablefaro-macrostomia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de ablefaro-macrostomia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Infantile epileptic spasms syndrome as a new phenotype in TOP2B deficiency caused by a de novo variant: a case report and literature review.
- Upper and lower eyelid contour and positional changes after deep skin grafts in ablepharon macrostomia syndrome.
- Modified Reverse Hatchet Flap for Ablepharon-Macrostomia Syndrome.
- Cryptophthalmos: associated syndromes and genetic disorders.
- Ocular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
- Ablepharon macrostomia syndrome: Absent prepuce in the first case report in West Africa.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:920(Orphanet)
- OMIM OMIM:200110(OMIM)
- MONDO:0008693(MONDO)
- GARD:3(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508585(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
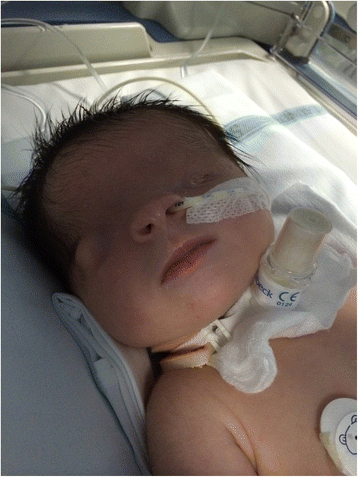
Síndrome de ablefaro-macrostomia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata