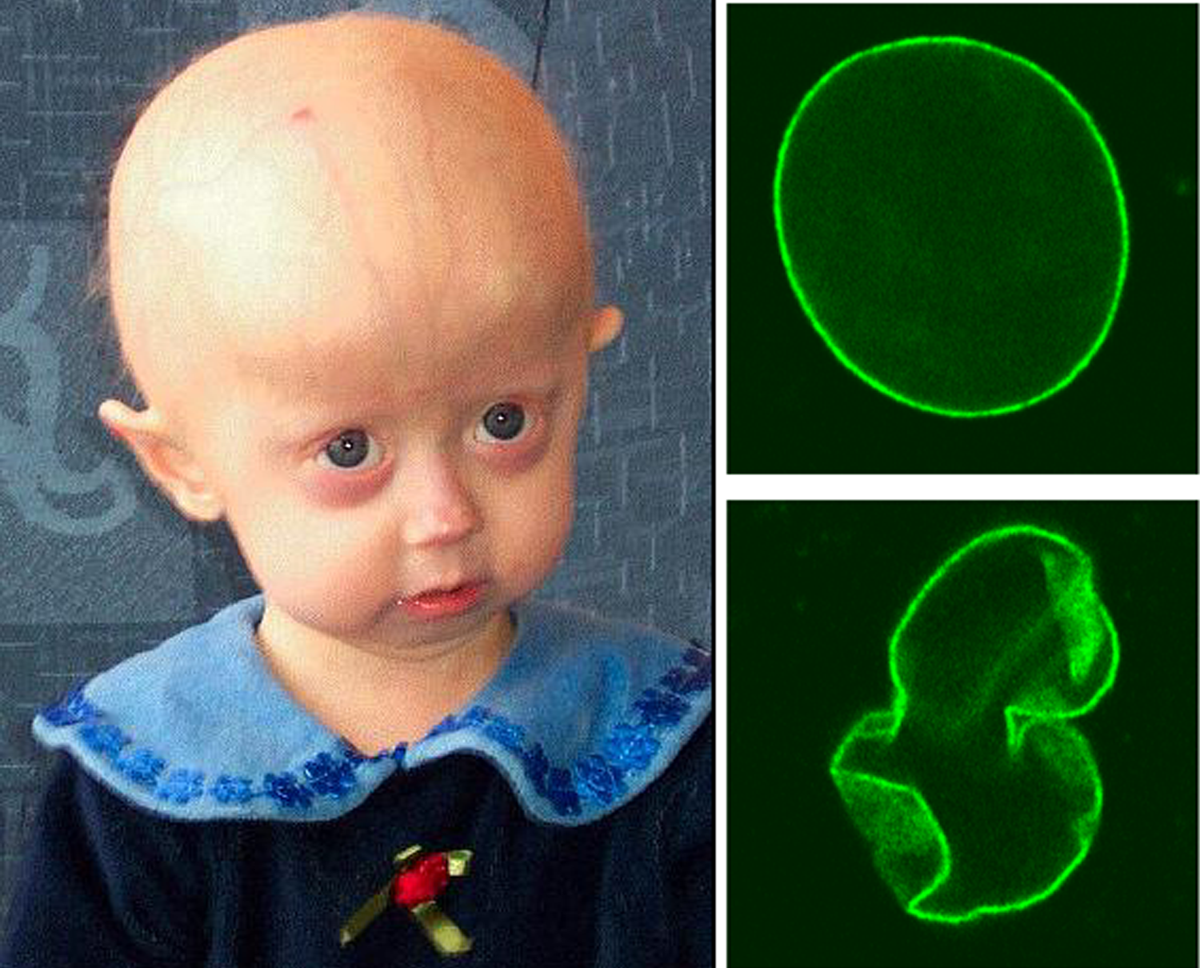
Doença genética autossômica recessiva rara caracterizada por dismorfismo facial (fissuras palpebrais inclinadas para baixo, ponte nasal larga e plana e boca pequena) com aparência progeróide, fontanela grande e de fechamento tardio, cútis laxa (CL), hiperfrouxidão articular, movimentos atetóides e hiperreflexia, retardo de crescimento pré e pós-natal, déficit intelectual e atraso no desenvolvimento, e turvação da córnea e catarata.
Introdução
O que você precisa saber de cara
Doença genética autossômica recessiva rara caracterizada por dismorfismo facial (fissuras palpebrais inclinadas para baixo, ponte nasal larga e plana e boca pequena) com aparência progeróide, fontanela grande e de fechamento tardio, cútis laxa (CL), hiperfrouxidão articular, movimentos atetóides e hiperreflexia, retardo de crescimento pré e pós-natal, déficit intelectual e atraso no desenvolvimento, e turvação da córnea e catarata.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 36 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 108 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Bifunctional enzyme that converts glutamate to glutamate 5-semialdehyde, an intermediate in the biosynthesis of proline, ornithine and arginine
MitochondrionMitochondrion matrix
Cutis laxa, autosomal recessive, 3A
A syndrome characterized by facial dysmorphism with a progeroid appearance, large and late-closing fontanel, cutis laxa, joint hyperlaxity, athetoid movements and hyperreflexia, pre- and postnatal growth retardation, intellectual deficit, developmental delay, and ophthalmologic abnormalities.
Oxidoreductase that catalyzes the last step in proline biosynthesis, which corresponds to the reduction of pyrroline-5-carboxylate to L-proline using NAD(P)H (PubMed:16730026, PubMed:19648921, PubMed:23024808, PubMed:28258219). At physiologic concentrations, has higher specific activity in the presence of NADH (PubMed:16730026, PubMed:23024808). Involved in the cellular response to oxidative stress (PubMed:16730026, PubMed:19648921)
Mitochondrion
Cutis laxa, autosomal recessive, 2B
A disorder characterized by an excessive congenital skin wrinkling, a large fontanelle with delayed closure, a typical facial appearance with downslanting palpebral fissures, a general connective tissue weakness, and varying degrees of growth and developmental delay and neurological abnormalities. Patients do not manifest metabolic abnormalities.
Variantes genéticas (ClinVar)
328 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 697 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome De Barsy
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Two novel homozygous variants of ATP6V0A2 and ALDH18A1 lead to autosomal recessive cutis laxa type 2 and 3 in two Pakistani families.
Autosomal recessive cutis laxa type 2A (ARCL2A; OMIM: 219200) is characterized by neurovegetative, developmental and progeroid elastic skin anomalies. It is caused by biallelic variation in ATPase, H+ transporting V0 subunit A2 (ATP6V0A2; OMIM: 611716) located on chromosome 12q24.31. Autosomal recessive cutis laxa type 3A (ARCL3A; OMIM: 219150) is another subclinical type characterized by short stature, ophthalmological abnormalities and a progeria-like appearance. The ARCL3A is caused by loss of function alterations in the aldehyde dehydrogenase 18 family member A1 (ALDH18A1; OMIM: 138250) gene located at chromosome 10q24.1. Whole-exome sequencing (WES), and Sanger sequencing were performed for molecular diagnosis. 3D protein modeling was performed to investigate the deleterious effect of the variant on protein structure. In this study, clinical and molecular diagnosis were performed for two families, ED-01 and DWF-41, which displayed hallmark features of ARCL2A and ARCL3A, respectively. Three affected individuals in the ED-01 family (IV-4, IV-5 and V-3) displayed sagging loose skin, down-slanting palpebral fissures, excessive wrinkles on the abdomen, hands and feet, and prominent veins on the trunk. Meanwhile the affected individuals in the DWF-41 family (V-2 and V-3) had progeroid skin, short stature, dysmorphology, low muscle tone, epilepsy, lordosis, scoliosis, delayed puberty and internal genitalia. WES in the index patient (ED-01: IV-4) identified a novel homozygous deletion (NM_012463.3: c.1977_1980del; p.[Val660LeufsTer23]) in exon 16 of the ATP6V0A2 while in DWF-41 a novel homozygous missense variant (NM_001323413.1:c.1867G>A; p.[Asp623Asn]) in exon 15 of the ALDH18A1 was identified. Sanger validation in all available family members confirmed the autosomal recessive modes of inheritances in each family. Three dimensional in-silico protein modeling suggested deleterious impact of the identified variants. Furthermore, these variants were assigned class 1 or "pathogenic" as per guidelines of American College of Medical Genetics 2015. Screening of ethnically matched healthy controls (n = 200 chromosomes), excluded the presence of these variations in general population. To the best of our knowledge, this is the first report of ATP6V0A2 and ALDH18A1 variations in the Pakhtun ethnicity of Pakistani population. The study confirms that WES can be used as a first-line diagnostic test in patients with cutis laxa, and provides basis for population screening and premarital testing to reduce the diseases burden in future generations.
De Barsy Syndrome: A Case Report of a Rare Genetic Disorder.
De Barsy syndrome (DBS) is an exceedingly rare autosomal recessively inherited genetic disorder that manifests as premature aging with progeroid features. Typically, the skin loses its elasticity, causing laxity, wrinkling, and sagging. Other characteristics include ophthalmological, orthopedic, and neurological abnormalities. As of 2011, only 27 DBS cases had been recorded. This paper reports the case of a two-day-old female infant who was referred to the pediatrics department with complaints of lax skin, a progeroid appearance, a short stature, hazy corneas, and bilateral claw-like hands with thin overlapping fingers. She also had features of pectus excavatum and visible veins over her chest and abdomen. There was a history of third-degree consanguineous parents in this patient. This patient was diagnosed with De Barsy syndrome due to findings on the Verhoeff Van Gieson staining, which demonstrated a marked decrease in elastic tissue fibers. Palliative care was recommended for this infant. We report this case considering its extreme rarity.
Progeroid syndrome of De Barsy - a case report and review of ophthalmic literature.
This report describes a very rare case of progeroid syndrome of De Barsy (Cutis laxa-corneal clouding syndrome). A 2 year-old child presented to the pediatric ophthalmology outpatients with bilateral congenital corneal opacification along with dysmorphic facial features, including loose wrinkled skin, progeroid appearance, delayed milestones, short stature, multiple hyper-extensible joints, muscular hypotonia, pectus excavatum and congenital dislocation of the hip joint. The child underwent a detailed ophthalmic work up and systemic evaluation by a clinical geneticist. Ophthalmic management in the form of bilateral sequential penetrating keratoplasties and a left eye trabeculectomy for medically uncontrolled angle-closure glaucoma was performed. Visual rehabilitation with glasses and amblyopia therapy is ongoing. Histopathology of the corneal button revealed loss of the bowman's layer which was replaced by a fibrous pannus while the stroma showed loss of stromal lamellar architecture with anterior and mid stroma showing vascularization. Genetic testing confirmed a mutation in the PYCR1 gene for a homozygous autosomal recessive cutis laxa type IIB. Although rare, De Barsy syndrome is an important cause of corneal opacification at birth with multiple systemic abnormalities that requires intervention.
Clinical and Molecular Delineation of Cutis Laxa Syndromes: Paradigms for Homeostasis.
Cutis laxa (CL) syndromes are a large and heterogeneous group of rare connective tissue disorders that share loose redundant skin as a hallmark clinical feature, which reflects dermal elastic fiber fragmentation. Both acquired and congenital-Mendelian- forms exist. Acquired forms are progressive and often preceded by inflammatory triggers in the skin, but may show systemic elastolysis. Mendelian forms are often pleiotropic in nature and classified upon systemic manifestations and mode of inheritance. Though impaired elastogenesis is a common denominator in all Mendelian forms of CL, the underlying gene defects are diverse and affect structural components of the elastic fiber or impair metabolic pathways interfering with cellular trafficking, proline synthesis, or mitochondrial functioning. In this chapter we provide a detailed overview of the clinical and molecular characteristics of the different cutis laxa types and review the latest insights on elastic fiber assembly and homeostasis from both human and animal studies.
A proposal of rehabilitative approach in the rare disease "De Barsy Syndrome": case report.
De Barsy syndrome is an autosomal recessive condition characterized by an progeroid appearance with distinctive facial features and cutis laxa. Ophthalmological, orthopedic, and neurological anomalies are generally also present. This syndrome is rare and the complex therapeutic management, from a surgical but also rehabilitative point of view, has not been recognized. The aim of this paper is to describe a possible rehabilitative protocol, after an orthopedic surgical treatment, in a child with De Barsy Syndrome. A 6-year-old boy was born with a congenital bilateral hip dysplasia associated with bilateral congenital foot deformity (vertical talus). Moreover, he showed stereotypic dyskinetic movements and psychomotor delay with cognitive impairment and absent language; the sitting position was maintained with orthoses to support the trunk control and the standing position was not acquired. He was treated with pinstripe knee-highs for the foot and double nappy for the hips. At 19 months old, he underwent a two stage surgical approach for a bilateral pronated valgus foot with severe talonavicular subluxation. Satisfactory hip range of motion was achieved by conservative treatment alone. Afterwards, for the foot laxity and the flat-pronated foot corrective shoes were prescribed. The main rehabilitative goals were: attention improvement, visual exploration for foot-eye and hand-eye coordination, encourage the essential prerequisites of language, controlling the upright position using support, improving hip-knee-foot relationship, improving load transfer between the right and left sides of the body, and bimanual coordination. The rehabilitation process lasted six months, three times a week, for a time from 30 minutes to 60 minutes per session. The results were encouraging and the patient acquired the possibility of sitting with the indicated postural system, the possibility of assuming an upright position and taking a few steps with the aid of rollator with a postural stabilization system for the pelvis.
Publicações recentes
De Barsy Syndrome: A Case Report of a Rare Genetic Disorder.
Progeroid syndrome of De Barsy - a case report and review of ophthalmic literature.
Clinical and Molecular Delineation of Cutis Laxa Syndromes: Paradigms for Homeostasis.
A proposal of rehabilitative approach in the rare disease "De Barsy Syndrome": case report.
Clinical implications of de Barsy syndrome.
📚 EuropePMC22 artigos no totalmostrando 9
Two novel homozygous variants of ATP6V0A2 and ALDH18A1 lead to autosomal recessive cutis laxa type 2 and 3 in two Pakistani families.
The journal of gene medicineDe Barsy Syndrome: A Case Report of a Rare Genetic Disorder.
CureusProgeroid syndrome of De Barsy - a case report and review of ophthalmic literature.
Ophthalmic geneticsClinical and Molecular Delineation of Cutis Laxa Syndromes: Paradigms for Homeostasis.
Advances in experimental medicine and biologyA proposal of rehabilitative approach in the rare disease "De Barsy Syndrome": case report.
La Clinica terapeuticaClinical implications of de Barsy syndrome.
Paediatric anaesthesiaDe Barsy syndrome type B presenting with cardiac and genitourinary abnormalities.
Clinical dysmorphologyA 5-year Journey with Cutis Laxa in an Indian Child: The De Barsy Syndrome Revisited.
Indian journal of dermatologyRecurrent De Novo Mutations Affecting Residue Arg138 of Pyrroline-5-Carboxylate Synthase Cause a Progeroid Form of Autosomal-Dominant Cutis Laxa.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome De Barsy.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome De Barsy
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Two novel homozygous variants of ATP6V0A2 and ALDH18A1 lead to autosomal recessive cutis laxa type 2 and 3 in two Pakistani families.
- De Barsy Syndrome: A Case Report of a Rare Genetic Disorder.
- Progeroid syndrome of De Barsy - a case report and review of ophthalmic literature.
- Clinical and Molecular Delineation of Cutis Laxa Syndromes: Paradigms for Homeostasis.
- A proposal of rehabilitative approach in the rare disease "De Barsy Syndrome": case report.
- Clinical implications of de Barsy syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2962(Orphanet)
- MONDO:0017569(MONDO)
- GARD:49(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3961699(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome De Barsy
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata