A síndrome RAPADILINO é uma condição cujo nome é uma sigla que indica seus principais sinais: * **RA**: para um defeito no osso rádio (um dos ossos do antebraço). * **PA**: para rótulas (ossos do joelho) que são pequenas ou ausentes, e para uma fenda ou um céu da boca muito arqueado. * **DI**: para diarreia e articulações que se deslocam com facilidade. * **LI**: para baixa estatura (ser pequeno) e malformações (problemas na formação) nos braços e pernas. * **NO**: para um nariz longo e fino, e inteligência normal.
Introdução
O que você precisa saber de cara
A síndrome RAPADILINO é uma condição cujo nome é uma sigla que indica seus principais sinais: * **RA**: para um defeito no osso rádio (um dos ossos do antebraço). * **PA**: para rótulas (ossos do joelho) que são pequenas ou ausentes, e para uma fenda ou um céu da boca muito arqueado. * **DI**: para diarreia e articulações que se deslocam com facilidade. * **LI**: para baixa estatura (ser pequeno) e malformações (problemas na formação) nos braços e pernas. * **NO**: para um nariz longo e fino, e inteligência normal.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 8 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 31 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisAn ATP-dependent DNA helicase which unwinds dsDNA with a 3'-overhang in a 3'-5' direction (PubMed:28653661). Does not unwind more than 18 bp of dsDNA (PubMed:28653661). May modulate chromosome segregation. The N-terminal domain (residues 1-54) binds DNA Y-shaped DNA better than ss- or dsDNA (PubMed:22730300). The core helicase domain binds ssDNA (PubMed:22730300, PubMed:28653661)
CytoplasmNucleus
RAPADILINO syndrome
Disease characterized by radial and patellar aplasia or hypoplasia.
Variantes genéticas (ClinVar)
688 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 172 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome RAPADILINO
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
RECQL4 encodes a RecQ helicase, one of a family of DNA unwinding enzymes with roles in DNA replication, double-strand break repair, and genomic stability. Pathogenic variants in RECQL4 are clinically associated with 3 rare autosomal recessive conditions: Rothmund-Thomson syndrome type II, Baller-Gerold syndrome, and RAPADILINO syndrome. These 3 syndromes show overlapping growth retardation, low bone density, and skeletal defects affecting the arms and hands. Here, we take advantage of the ability to generate one-sided CRISPR knockdowns of recql4 in Xenopus laevis tadpoles. Tadpoles develop normally until feeding starts, after which growth slows on the edited side, leading to a curved posture, smaller eyes (microphthalmia), and reduced head size (microcephaly). Forelimb buds fail to develop, leading to complete absence of the forelimb on the edited side. Additionally, Meckel's cartilage (lower jaw) ossification is absent or reduced and the hyoid cartilage is smaller, but this is not due to deficiencies in cranial neural crest migration on the edited side. Knockdown of recql4 also results in hypoplastic vasculature, with reduced branching from the aorta on the edited side. Taken together, our results clearly show the utility of unilateral CRISPR editing in Xenopus for understanding the specific phenotypic developmental effects of mutations affecting cell proliferation.
Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
Baller-Gerold syndrome (BGS, OMIM: 218600), RAPADILINO syndrome (OMIM 266280), and Rothmund-Thomson syndrome (RTS, OMIM 266280), which are caused in some cases by RECQL4 pathogenic variants, show autosomal recessive inheritance. Some refer to them collectively as RECQL4 syndromes. Most cases have been reported during infancy and childhood periods. However, there have been no reports of phenotypes resulting in a lethal course in the perinatal period. We identified two fetuses with biallelic RECQL4 pathogenic variants during the perinatal period. The two fetuses with RECQL4 syndrome showed structural abnormalities, including severely hypoplastic forearms and lower legs. One fetus also had severe pulmonary hypoplasia. One case resulted in neonatal death because of respiratory failure, and the other was artificially terminated during pregnancy. The RECQL4 pathogenic variants were identified by exome sequencing followed by Sanger sequencing. The biallelic RECQL4 pathogenic variants can induce a lethal skeletal disorder.
Human RecQ Helicases in DNA Double-Strand Break Repair.
RecQ DNA helicases are a conserved protein family found in bacteria, fungus, plants, and animals. These helicases play important roles in multiple cellular functions, including DNA replication, transcription, DNA repair, and telomere maintenance. Humans have five RecQ helicases: RECQL1, Bloom syndrome protein (BLM), Werner syndrome helicase (WRN), RECQL4, and RECQL5. Defects in BLM and WRN cause autosomal disorders: Bloom syndrome (BS) and Werner syndrome (WS), respectively. Mutations in RECQL4 are associated with three genetic disorders, Rothmund-Thomson syndrome (RTS), Baller-Gerold syndrome (BGS), and RAPADILINO syndrome. Although no genetic disorders have been reported due to loss of RECQL1 or RECQL5, dysfunction of either gene is associated with tumorigenesis. Multiple genetically independent pathways have evolved that mediate the repair of DNA double-strand break (DSB), and RecQ helicases play pivotal roles in each of them. The importance of DSB repair is supported by the observations that defective DSB repair can cause chromosomal aberrations, genomic instability, senescence, or cell death, which ultimately can lead to premature aging, neurodegeneration, or tumorigenesis. In this review, we will introduce the human RecQ helicase family, describe in detail their roles in DSB repair, and provide relevance between the dysfunction of RecQ helicases and human diseases.
Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells.
Human RecQ helicases that share homology with E. coli RecQ helicase play critical roles in diverse biological activities such as DNA replication, transcription, recombination and repair. Mutations in three of the five human RecQ helicases (RecQ1, WRN, BLM, RecQL4 and RecQ5) result in autosomal recessive syndromes characterized by accelerated aging symptoms and cancer incidence. Mutational inactivation of Werner (WRN) and Bloom (BLM) genes results in Werner syndrome (WS) and Bloom syndrome (BS) respectively. However, mutations in RecQL4 result in three human disorders: (I) Rothmund-Thomson syndrome (RTS), (II) RAPADILINO and (III) Baller-Gerold syndrome (BGS). Cells from WS, BS and RTS are characterized by a unique chromosomal anomaly indicating that each of the RecQ helicases performs specialized function(s) in a non-redundant manner. Elucidating the biological functions of RecQ helicases will enable us to understand not only the aging process but also to determine the cause for age-associated human diseases. Recent biochemical and molecular studies have given new insights into the multifaceted roles of RecQL4 that range from genomic stability to carcinogenesis and beyond. This review summarizes some of the existing and emerging knowledge on diverse biological functions of RecQL4 and its significance as a potential molecular target for cancer therapy.
The DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation.
RECQL4 mutations are associated with Rothmund Thomson Syndrome (RTS), RAPADILINO Syndrome and Baller-Gerold Syndrome. These patients display a range of benign skeletal abnormalities such as low bone mass. In addition, RTS patients have a highly increased incidence of osteosarcoma (OS). The role of RECQL4 in normal adult bone development and homeostasis is largely uncharacterized and how mutation of RECQL4 contributes to OS susceptibility is not known. We hypothesised that Recql4 was required for normal skeletal development and both benign and malignant osteoblast function, which we have tested in the mouse. Recql4 deletion in vivo at the osteoblastic progenitor stage of differentiation resulted in mice with shorter bones and reduced bone volume, assessed at 9 weeks of age. This was associated with an osteoblast intrinsic decrease in mineral apposition rate and bone formation rate in the Recql4-deficient cohorts. Deletion of Recql4 in mature osteoblasts/osteocytes in vivo, however, did not cause a detectable phenotype. Acute deletion of Recql4 in primary osteoblasts or shRNA knockdown in an osteoblastic cell line caused failed proliferation, accompanied by cell cycle arrest, induction of apoptosis and impaired differentiation. When cohorts of animals were aged long term, the loss of Recql4 alone was not sufficient to initiate OS. We then crossed the Recql4fl/fl allele to a fully penetrant OS model (Osx-Cre p53fl/fl). Unexpectedly, the Osx-Cre p53fl/flRecql4fl/fl (dKO) animals had a significantly increased OS-free survival compared to Osx-Cre p53fl/fl or Osx-Cre p53fl/flRecql4fl/+ (het) animals. The extended survival was explained when the Recql4 status in the tumors that arose was assessed, and in no case was there complete deletion of Recql4 in the dKO OS. These data provide a mechanism for the benign skeletal phenotypes of RECQL4 mutation syndromes. We propose that tumor suppression and osteosarcoma susceptibility are most likely a function of mutant, not null, alleles of RECQL4.
Publicações recentes
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
📖 RevisãoSevere Phenotype With RECQL4 Syndrome: A Report of Two Cases.
Human RecQ Helicases in DNA Double-Strand Break Repair.
Immunodeficiency in a Child with Rapadilino Syndrome: A Case Report and Review of the Literature.
The DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation.
📚 EuropePMC7 artigos no totalmostrando 7
Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
G3 (Bethesda, Md.)Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
American journal of medical genetics. Part AHuman RecQ Helicases in DNA Double-Strand Break Repair.
Frontiers in cell and developmental biologyHuman RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells.
Cancer lettersImmunodeficiency in a Child with Rapadilino Syndrome: A Case Report and Review of the Literature.
Case reports in immunologyThe DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation.
PLoS geneticsRECQL4 Regulates p53 Function In Vivo During Skeletogenesis.
Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral ResearchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome RAPADILINO.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome RAPADILINO
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unilateral loss of recql4 function in Xenopus laevis tadpoles leads to ipsilateral ablation of the forelimb, hypoplastic Meckel's cartilage, and vascular defects.
- Severe Phenotype With RECQL4 Syndrome: A Report of Two Cases.
- Human RecQ Helicases in DNA Double-Strand Break Repair.
- Human RecQL4 helicase plays multifaceted roles in the genomic stability of normal and cancer cells.
- The DNA helicase recql4 is required for normal osteoblast expansion and osteosarcoma formation.
- Immunodeficiency in a Child with Rapadilino Syndrome: A Case Report and Review of the Literature.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3021(Orphanet)
- OMIM OMIM:266280(OMIM)
- MONDO:0009955(MONDO)
- GARD:4637(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3508578(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
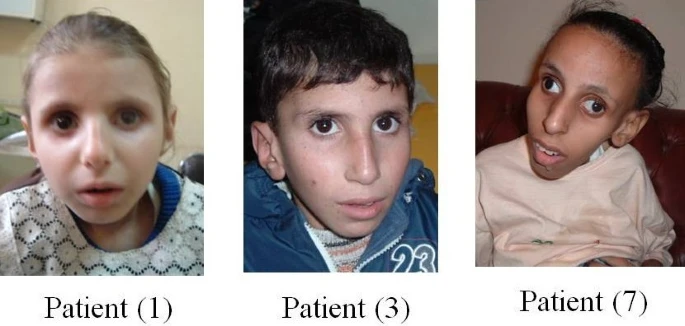
Síndrome RAPADILINO
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata