A deficiência de adenilosuccinato liase (deficiência de ADSL) é um distúrbio do metabolismo das purinas caracterizado por deficiência intelectual, atraso e/ou regressão psicomotora, convulsões e características autistas.
Introdução
O que você precisa saber de cara
A deficiência de adenilosuccinato liase (deficiência de ADSL) é um distúrbio do metabolismo das purinas caracterizado por deficiência intelectual, atraso e/ou regressão psicomotora, convulsões e características autistas.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 20 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCatalyzes two non-sequential steps in de novo AMP synthesis: converts (S)-2-(5-amino-1-(5-phospho-D-ribosyl)imidazole-4-carboxamido)succinate (SAICAR) to fumarate plus 5-amino-1-(5-phospho-D-ribosyl)imidazole-4-carboxamide, and thereby also contributes to de novo IMP synthesis, and converts succinyladenosine monophosphate (SAMP) to AMP and fumarate
Adenylosuccinase deficiency
An autosomal recessive disorder characterized by the accumulation in the body fluids of succinylaminoimidazole-carboxamide riboside (SAICA-riboside) and succinyladenosine (S-Ado). Most children display marked psychomotor delay, often accompanied by epilepsy or autistic features, or both, although some patients may be less profoundly retarded. Occasionally, growth retardation and muscular wasting are also present.
Variantes genéticas (ClinVar)
174 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 838 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência de adenilossuccinato liase
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
2 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Severe Prenatal Presentation of Adenylosuccinate Lyase Deficiency Caused by a Synonymous ADSL Variant Inducing Aberrant Splicing.
What is already known about this topic? ◦. ADSL deficiency is a rare metabolic disorder, typically diagnosed postnatally with variable severity. ◦. ADSL activity is known to be reduced in affected patients. ◦. Pathogenic variants in ADSL are predominantly missense or truncating; no pathogenic synonymous variants have been reported. What does this study add? ◦. The first functionally confirmed case of severe prenatal‐onset ADSL deficiency is described. ◦. The first pathogenic synonymous ADSL variant causing aberrant splicing is identified. ◦. Reduced ADSL activity in PBMCs of unaffected parents is demonstrated for the first time.
Allopurinol Treatment Improves Cognitive Skills, Adaptive Behavior, and Biochemical Markers in Young Patients With Adenylosuccinate Lyase Deficiency.
Adenylosuccinate lyase deficiency (ADSLD) is a rare neurological disorder characterized by psychomotor retardation, autistic behaviors, and seizures, with no specific treatment available. ADSL catalyzes the transformation of succinylaminoimidazole carboxamide ribotide (SAICAr) to AICAR, and succinyl-AMP (S-AMP) to AMP. The pathogenesis of the disease is primarily attributed to the toxicity of elevated SAICAr concentrations. Allopurinol, used primarily for hyperuricemia, inhibits purine synthesis and may reduce SAICAr levels. We hypothesized that administering allopurinol could decrease SAICAr levels and lead to clinical improvement. A Phase II, prospective trial evaluated the efficacy of allopurinol in patients with ADSLD over 12 months. Eight participants (four children, four young adults) with developmental delay and high SAICAr levels received Zyloric (10-20 mg/kg/day, maximum 400 mg/day for children and 900 mg/day for adults). The study assessed changes in adaptive and cognitive functioning, behavior, and urinary levels of SAICAr and succinyl-adenosine (S-Ado). Results showed clinical improvements in younger, less cognitively impaired patients, indicated by better Vineland Adaptive Behavior Scale (VABS II) scores and reduced hyperactivity on the Aberrant Behavior Checklist (ABC) and Conners Rating Scale-Revised (CRSR). These improvements correlated with significant decreases in urinary SAICAr levels and an increased S-Ado/SAICAr ratio. No changes were observed in older or noncompliant patients. Allopurinol had no effect on epilepsy but was well tolerated. Allopurinol showed behavioral and developmental benefits in younger ADSLD patients, suggesting that it may be a viable treatment option.
ADSL deficiency is a secondary mitochondrial disease affecting organelle homeostasis and ERK2/AKT signaling in a linear genotype-phenotype relation.
Adenylosuccinate lyase deficiency (ADSLd) is a rare autosomal recessive purine metabolism disorder with several clinical manifestations. While toxic substrate accumulation is a known hallmark, no additional molecular mechanisms have been established. Here, we show that ADSLd is associated with mitochondrial dysfunction, including increased fragmentation, impaired respiration, and reduced ATP production. The severity of mitochondrial impairment correlates with ADSLd pathology, especially in mitochondria-dependent tissues. We also identify defects in mitochondrial dynamics and transport linked to ERK2 and AKT suppression. Notably, overexpressing constitutively active ERK2 or supplementing purine intermediates partially rescues the mitochondrial phenotype. These findings suggest an alternative disease mechanism and highlight mitochondrial metabolism as a potential therapeutic target in ADSLd.
Emerging role of complement system in the induction of neuroinflammation in adenylosuccinate lyase deficiency disorder.
Adenylosuccinate lyase deficiency disorder (ADSLDD) is an ultra-rare autosomal recessive metabolic condition that leads to severe neurological impairment, with an estimated global prevalence of approximately 0.00125 cases per 100,000 individuals. Clinically, ADSLDD presents in three distinct phenotypes: the fatal neonatal form, the childhood form, and the more slowly progressive form, each characterized by varying degrees of developmental and neurological dysfunction. The disorder is caused by pathogenic variants in the ADSL gene, leading to impaired enzymatic activity and the accumulation of toxic substrates particularly succinyladenosine (S-Ado) and succinylaminoimidazole carboxamide riboside (SAICAr). The ratio of S-Ado to SAICAr in cerebrospinal fluid has been correlated with disease severity, where lower ratios are associated with more severe clinical outcomes. However, the precise mechanisms linking elevated SAICAr levels to neurological damage remain incompletely understood. This review summarizes current insights into the metabolic dysfunction and immune activation observed in ADSLDD, with a focus on the role of SAICAr in promoting neuroinflammation. We highlight emerging hypotheses implicating activation of the alternative complement pathway as a key driver of inflammation, blood-brain barrier disruption, and progressive neurodegeneration. By synthesizing recent findings, this review underscores the urgent need for mechanistic studies and therapeutic exploration, particularly targeting complement activation, as a promising strategy to mitigate inflammation and improve clinical outcomes in ADSLDD.
Case Report: Adenylosuccinate lyase deficiency type I caused by splicing disruption due to a novel missense variant in the ADSL gene.
Adenylosuccinate lyase deficiency (ALD) is a rare neurometabolic disorder caused by biallelic loss-of-function variants in the ADSL gene. We report a severe type I ALD case involving a 2-year-old boy presenting with early-onset polymorphic seizures (clonic/myoclonic), developmental delay, and progressive neurological deterioration. Seizures were temporarily controlled with ethosuximide and vigabatrin, though neurodegeneration progressed. Analysis of whole-exome sequencing data revealed compound-heterozygous variants in the ADSL gene: the known pathogenic missense variant c.340T>C (p.Tyr114His) and a novel variant c.859A>G (p.Ile287Val). Although p.Ile287Val is predicted to be benign at the protein level, RNA analysis demonstrated that c.859A>G activates a cryptic splice site in exon 8, resulting in aberrant transcripts (64%, 4-bp deletion, targeted by nonsense-mediated decay) and a smaller proportion of normal transcripts (36%) encoding the p.Ile287Val protein. This case highlights splicing disruption as a novel pathogenic mechanism in ALD and expands the mutational spectrum associated with the disease. This case also underscores the importance of integrating RNA analysis with genomic data to uncover cryptic splicing defects, especially when protein-level predictions suggest benignity.
Publicações recentes
Severe Prenatal Presentation of Adenylosuccinate Lyase Deficiency Caused by a Synonymous ADSL Variant Inducing Aberrant Splicing.
Case Report: Adenylosuccinate lyase deficiency type I caused by splicing disruption due to a novel missense variant in the ADSL gene.
Allopurinol Treatment Improves Cognitive Skills, Adaptive Behavior, and Biochemical Markers in Young Patients With Adenylosuccinate Lyase Deficiency.
🥈 ObservacionalADSL deficiency is a secondary mitochondrial disease affecting organelle homeostasis and ERK2/AKT signaling in a linear genotype-phenotype relation.
Emerging role of complement system in the induction of neuroinflammation in adenylosuccinate lyase deficiency disorder.
📚 EuropePMC64 artigos no totalmostrando 28
Severe Prenatal Presentation of Adenylosuccinate Lyase Deficiency Caused by a Synonymous ADSL Variant Inducing Aberrant Splicing.
Prenatal diagnosisCase Report: Adenylosuccinate lyase deficiency type I caused by splicing disruption due to a novel missense variant in the ADSL gene.
Frontiers in geneticsAllopurinol Treatment Improves Cognitive Skills, Adaptive Behavior, and Biochemical Markers in Young Patients With Adenylosuccinate Lyase Deficiency.
Journal of inherited metabolic diseaseADSL deficiency is a secondary mitochondrial disease affecting organelle homeostasis and ERK2/AKT signaling in a linear genotype-phenotype relation.
Cell reportsEmerging role of complement system in the induction of neuroinflammation in adenylosuccinate lyase deficiency disorder.
Brain, behavior, & immunity - healthElectroclinical features and phenotypic differences in adenylosuccinate lyase deficiency: Long-term follow-up of seven patients from four families and appraisal of the literature.
Epilepsia openAdenylosuccinate lyase deficiency affects neurobehavior via perturbations to tyramine signaling in Caenorhabditis elegans.
PLoS geneticsA Caenorhabditis elegans model of adenylosuccinate lyase deficiency reveals neuromuscular and reproductive phenotypes of distinct etiology.
Molecular genetics and metabolismChild with adenylosuccinate lyase deficiency caused by a novel complex heterozygous mutation in the ADSL gene: A case report.
World journal of clinical casesAnticipatory banking of samples enables diagnosis of adenylosuccinase deficiency following molecular autopsy in an infant with vacuolating leukoencephalopathy.
American journal of medical genetics. Part APathway-specific effects of ADSL deficiency on neurodevelopment.
eLifeDisorders of purine biosynthesis metabolism.
Molecular genetics and metabolismClinical and molecular characterization of patients with adenylosuccinate lyase deficiency.
Orphanet journal of rare diseasesA newborn case of adenylosuccinate lyase deficiency with a novel heterozygous mutation diagnosed by whole exome sequencing.
Clinical neurology and neurosurgeryRegulation of purine metabolism connects KCTD13 to a metabolic disorder with autistic features.
iScienceMyoclonic tremor status as a presenting symptom of adenylosuccinate lyase deficiency.
European journal of medical geneticsADSL Deficiency - The Lesser-Known Metabolic Epilepsy in Infancy.
Indian journal of pediatricsVery mild isolated intellectual disability caused by adenylosuccinate lyase deficiency: a new phenotype.
Molecular genetics and metabolism reportsPAICS deficiency, a new defect of de novo purine synthesis resulting in multiple congenital anomalies and fatal outcome.
Human molecular geneticsBroadening phenotype of adenylosuccinate lyase deficiency: A novel clinical pattern resembling neuronal ceroid lipofuscinosis.
Molecular genetics and metabolism reportsCase 3: The Hypothermic Newborn.
NeoReviewsMRI findings of hypomyelination in adenylosuccinate lyase deficiency.
Radiology case reportsA mild form of adenylosuccinate lyase deficiency in absence of typical brain MRI features diagnosed by whole exome sequencing.
Italian journal of pediatricsNovel mutations in ADSL for Adenylosuccinate Lyase Deficiency identified by the combination of Trio-WES and constantly updated guidelines.
Scientific reportsPrevalence of adenylosuccinate lyase deficiency based on aggregated exome data.
Molecular genetics and metabolism reportsCRISPR-Cas9 induced mutations along de novo purine synthesis in HeLa cells result in accumulation of individual enzyme substrates and affect purinosome formation.
Molecular genetics and metabolismDiagnosis of adenylosuccinate lyase deficiency by metabolomic profiling in plasma reveals a phenotypic spectrum.
Molecular genetics and metabolism reportsThe nonessentiality of essential genes in yeast provides therapeutic insights into a human disease.
Genome researchAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência de adenilossuccinato liase.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência de adenilossuccinato liase
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Severe Prenatal Presentation of Adenylosuccinate Lyase Deficiency Caused by a Synonymous ADSL Variant Inducing Aberrant Splicing.
- Allopurinol Treatment Improves Cognitive Skills, Adaptive Behavior, and Biochemical Markers in Young Patients With Adenylosuccinate Lyase Deficiency.
- ADSL deficiency is a secondary mitochondrial disease affecting organelle homeostasis and ERK2/AKT signaling in a linear genotype-phenotype relation.
- Emerging role of complement system in the induction of neuroinflammation in adenylosuccinate lyase deficiency disorder.
- Case Report: Adenylosuccinate lyase deficiency type I caused by splicing disruption due to a novel missense variant in the ADSL gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:46(Orphanet)
- OMIM OMIM:103050(OMIM)
- MONDO:0007068(MONDO)
- GARD:550(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4682317(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
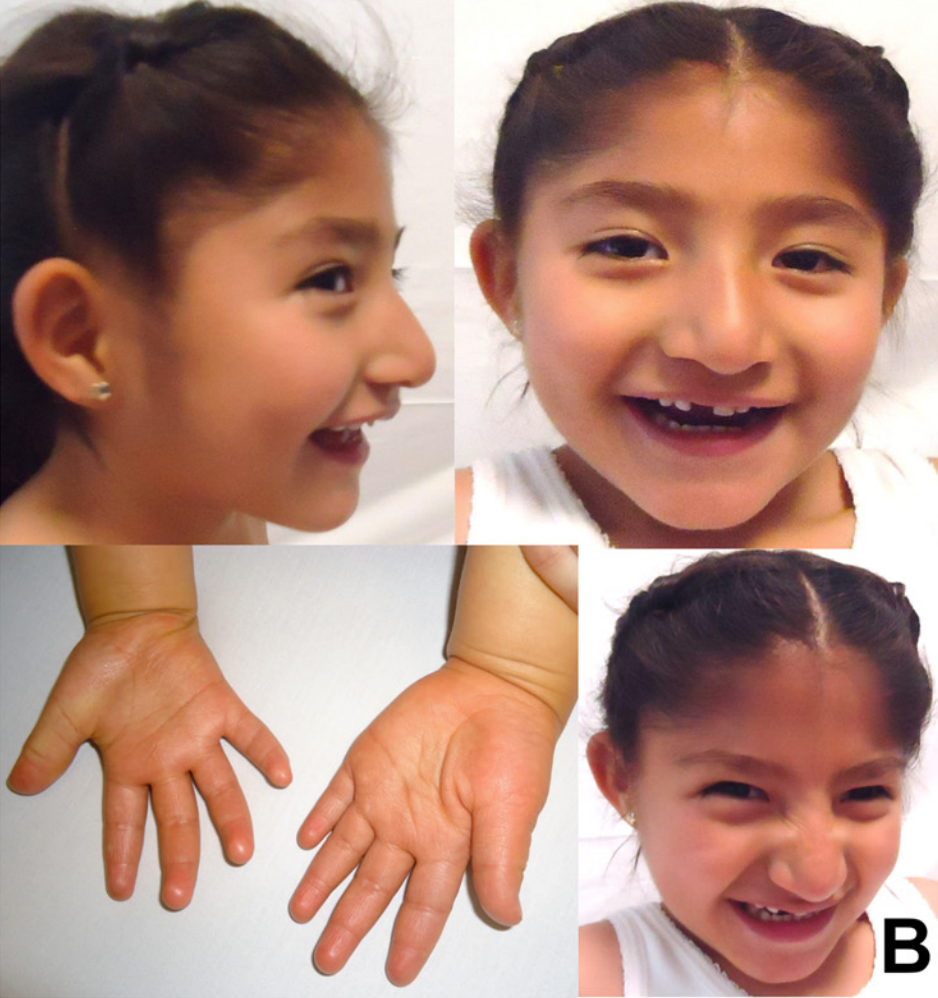
Deficiência de adenilossuccinato liase
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov