A Deficiência Sistêmica Primária de Carnitina (DSPC) é uma doença que pode ser fatal e que afeta o processo de queima de gorduras para gerar energia no corpo. Ela se manifesta tipicamente na primeira infância, com problemas no músculo do coração (cardiomiopatia), frequentemente acompanhada de fraqueza, músculos flácidos (hipotonia), dificuldade para crescer e se desenvolver, e crises convulsivas repetidas causadas por baixo açúcar no sangue e pela dificuldade do corpo em usar a gordura como fonte de energia, podendo levar ao coma.
Introdução
O que você precisa saber de cara
A Deficiência Sistêmica Primária de Carnitina (DSPC) é uma doença que pode ser fatal e que afeta o processo de queima de gorduras para gerar energia no corpo. Ela se manifesta tipicamente na primeira infância, com problemas no músculo do coração (cardiomiopatia), frequentemente acompanhada de fraqueza, músculos flácidos (hipotonia), dificuldade para crescer e se desenvolver, e crises convulsivas repetidas causadas por baixo açúcar no sangue e pela dificuldade do corpo em usar a gordura como fonte de energia, podendo levar ao coma.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 42 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisSodium-ion dependent, high affinity carnitine transporter. Involved in the active cellular uptake of carnitine. Transports one sodium ion with one molecule of carnitine (PubMed:10454528, PubMed:10525100, PubMed:10966938, PubMed:17509700, PubMed:20722056, PubMed:33124720). Also transports organic cations such as tetraethylammonium (TEA) without the involvement of sodium. Relative uptake activity ratio of carnitine to TEA is 11.3 (PubMed:10454528, PubMed:10525100, PubMed:10966938). In intestinal e
Cell membraneApical cell membraneBasal cell membraneEndoplasmic reticulum
Systemic primary carnitine deficiency
Autosomal recessive disorder of fatty acid oxidation caused by defective carnitine transport. Present early in life with hypoketotic hypoglycemia and acute metabolic decompensation, or later in life with skeletal myopathy or cardiomyopathy.
Variantes genéticas (ClinVar)
430 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Deficiência de carnitina primária sistêmica
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
13 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Clinical, Biochemical and Molecular Characterisation of Newborns With Fatty Acid β-Oxidation Disorders: Novel Variants in the ACADM , ACADVL and SLC22A5 Genes.
In this study, we aimed to assess clinical, laboratory and molecular features of newborns with clinical suspicion for systemic primary carnitine deficiency (CUD), medium-chain acyl-CoA dehydrogenase deficiency (MCADD) and very long-chain acyl-CoA dehydrogenase deficiency (VLCADD). The implementation of newborn screening programs for fatty acid β-oxidation disorders (FAODs) has changed the natural course of these diseases, facilitating the initiation of preventive or therapeutic measures for affected newborns shortly after birth. This study included 94 newborns who were admitted between 2016 and 2023 because of biochemical signs of CUD, MCADD and VLCADD, and provided clinical, biochemical and genotypic data. Definitive molecular diagnosis confirmed that 16/94 newborns (17%) were true positives of the NBS, and 17 novel variants were detected in SLC22A5, ACADM and ACADVL genes. We assessed the clinical evolution of patients over time. This study expands the genotypic spectrum of SLC22A5, ACADM and ACADVL and highlights the role of genetics in identifying and correctly characterising FAODs.
Structural basis of sodium ion-dependent carnitine transport by OCTN2.
Carnitine is essential for the import of long-chain fatty acids into mitochondria, where they are used for energy production. The carnitine transporter OCTN2 (novel organic cation transporter 2, SLC22A5) mediates carnitine uptake across the plasma membrane and as such facilitates fatty acid metabolism in most tissues. OCTN2 dysfunction causes systemic primary carnitine deficiency (SPCD), a potentially lethal disorder. Despite its importance in metabolism, the mechanism of high-affinity, sodium ion-dependent transport by OCTN2 is unclear. Here we report cryo-EM structures of human OCTN2 in three conformations: inward-facing ligand-free, occluded carnitine- and Na+-bound, and inward-facing ipratropium-bound. These structures define key interactions responsible for carnitine transport and identify an allosterically coupled Na+ binding site housed within an aqueous cavity, separate from the carnitine-binding site. Combined with electrophysiology data, we provide a framework for understanding variants associated with SPCD and insight into how OCTN2 functions as the primary human carnitine transporter.
Systemic Primary Carnitine Deficiency Presenting With Substantia Nigra and Basal Ganglia Injury: A Case Report.
Systemic primary carnitine deficiency (SPCD) is a rare congenital fatty acid metabolism disorder causing impaired β-oxidation and energy production, leading to hypoglycemia, metabolic encephalopathy, and sudden death. Early diagnosis and treatment, including L-carnitine supplementation and fasting avoidance, can improve prognosis. However, newborn screening (NBS) criteria differ by region, and standardized guidelines are lacking. This report presents a case of SPCD undetected by NBS, resulting in basal ganglia damage and dystonia due to metabolic decompensation. A 1-year-9-month-old girl with no abnormalities on NBS presented with impaired consciousness. She exhibited hypoketotic hypoglycemia, hyperammonemia, and myocardial hypertrophy. Suspecting a fatty acid metabolism disorder, L-carnitine and high-calorie infusion were initiated. Laboratory tests revealed markedly low serum total and free carnitine levels, and genetic analysis confirmed a homozygous SLC22A5 mutation. Brain MRI on day 7 revealed bilateral basal ganglia and substantia nigra abnormalities. The patient developed severe dystonia and respiratory failure, requiring ECMO management. L-DOPA was initiated on day 62, resulting in improvements in dystonia, swallowing, and motor function. By day 88, MRI showed resolution of basal ganglia abnormalities, though cerebral atrophy persisted. Basal ganglia damage is a rare but severe SPCD complication. L-DOPA may alleviate dystonia by acting on dopaminergic neurons in the substantia nigra. Early ketone measurement during emergencies is crucial for diagnosing fatty acid metabolism disorders. A standardized NBS protocol with a defined carnitine cutoff value is essential for early detection and prevention of SPCD complications. Primary carnitine deficiency (PCD) is a disorder of the carnitine cycle that results in defective fatty acid oxidation. If untreated, it encompasses a broad clinical spectrum including: (1) metabolic decompensation in infancy typically presenting between age three months and two years with episodes of hypoketotic hypoglycemia, poor feeding, irritability, lethargy, hepatomegaly, elevated liver transaminases, and hyperammonemia triggered by fasting or common illnesses such as upper respiratory tract infection or gastroenteritis; (2) childhood myopathy involving heart and skeletal muscle with onset between age two and four years; (3) pregnancy-related decreased stamina or exacerbation of cardiac arrhythmia; (4) fatigability in adulthood; and (5) absence of symptoms. The latter two categories often include mothers diagnosed with PCD after newborn screening has identified low carnitine levels in their infants. The diagnosis of PCD is established in a proband with consistent biochemical analyte findings and/or suggestive clinical and laboratory features by identification of biallelic pathogenic variants in SLC22A5 on molecular genetic testing. In individuals with suspected PCD and negative molecular testing, a carnitine transport assay using cultured skin fibroblasts may be available. Targeted therapy: Metabolic decompensation and skeletal and cardiac muscle function improve with 100-200 mg/kg/day oral levocarnitine if it is started before irreversible organ damage occurs. Supportive care: Routine treatment includes preventing hypoglycemia with frequent feeding and avoidance of prolonged fasting; notifying designated metabolic center in advance of scheduled surgical or medical procedures; hospitalization for intravenous glucose administration for individuals who are required to fast for a procedure or who cannot tolerate oral intake due to illness such as gastroenteritis; implementing transitional care plan prior to adulthood. Emergency outpatient treatment includes levocarnitine and carbohydrate supplementation, antipyretics for fever, and antiemetics for occasional vomiting. Acute inpatient treatment includes high-calorie fluids, insulin as needed, intravenous or oral levocarnitine (100-200 mg/kg/day), evaluation of muscle and liver involvement by measuring serum creatine kinase concentration and liver transaminases, and evaluation by cardiologist with EKG and echocardiogram for cardiomyopathy. Surveillance: Monitor plasma carnitine concentration frequently until levels reach normal range; once levels reach normal range, measure three times a year during infancy and early childhood, twice a year in older children, and annually in adults. Assess growth and development at each visit throughout childhood. Neuropsychological testing and quality of life assessment as needed. EKG and echocardiogram annually during childhood and less frequently in adulthood. Agents/circumstances to avoid: Fasting longer than age-appropriate periods; catabolic illness; inadequate calorie provision during other stressors. Evaluation of relatives at risk: Evaluation of all sibs of any age by molecular genetic testing if the SLC22A5 pathogenic variants in the family are known or measurement of plasma-free carnitine concentration to identify as early as possible those who would benefit from institution of treatment and preventive measures. Pregnancy management: Pregnant women with PCD require close monitoring of plasma carnitine levels and increased carnitine supplementation as needed to maintain plasma carnitine levels in the normal range. PCD is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an SLC22A5 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the SLC22A5 pathogenic variants have been identified in an affected family member, molecular genetic carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
The Human OCTN Sub-Family: Gene and Protein Structure, Expression, and Regulation.
OCTN1 and OCTN2 are membrane transport proteins encoded by the SLC22A4 and SLC22A5 genes, respectively. Even though several transcripts have been predicted by bioinformatics for both genes, only one functional protein isoform has been described for each of them. Both proteins are ubiquitous, and depending on the physiopathological state of the cell, their expression is regulated by well-known transcription factors, although some aspects have been neglected. A plethora of missense variants with uncertain clinical significance are reported both in the dbSNP and the Catalogue of Somatic Mutations in Cancer (COSMIC) databases for both genes. Due to their involvement in human pathologies, such as inflammatory-based diseases (OCTN1/2), systemic primary carnitine deficiency (OCTN2), and drug disposition, it would be interesting to predict the impact of variants on human health from the perspective of precision medicine. Although the lack of a 3D structure for these two transport proteins hampers any speculation on the consequences of the polymorphisms, the already available 3D structures for other members of the SLC22 family may provide powerful tools to perform structure/function studies on WT and mutant proteins.
Harnessing Next-Generation Sequencing as a Timely and Accurate Second-Tier Screening Test for Newborn Screening of Inborn Errors of Metabolism.
In this study, we evaluated the implementation of a second-tier genetic screening test using an amplicon-based next-generation sequencing (NGS) panel in our laboratory during the period of 1 September 2021 to 31 August 2022 for the newborn screening (NBS) of six conditions for inborn errors of metabolism: citrullinemia type II (MIM #605814), systemic primary carnitine deficiency (MIM #212140), glutaric acidemia type I (MIM #231670), beta-ketothiolase deficiency (#203750), holocarboxylase synthetase deficiency (MIM #253270) and 3-hydroxy-3-methylglutaryl-CoA lyase deficiency (MIM # 246450). The custom-designed NGS panel can detect sequence variants in the relevant genes and also specifically screen for the presence of the hotspot variant IVS16ins3kb of SLC25A13 by the copy number variant calling algorithm. Genetic second-tier tests were performed for 1.8% of a total of 22,883 NBS samples. The false positive rate for these six conditions after the NGS second-tier test was only 0.017%, and two cases of citrullinemia type II would have been missed as false negatives if only biochemical first-tier testing was performed. The confirmed true positive cases were citrullinemia type II (n = 2) and systemic primary carnitine deficiency (n = 1). The false positives were later confirmed to be carrier of citrullinemia type II (n = 2), carrier of glutaric acidemia type I (n = 1) and carrier of systemic primary carnitine deficiency (n = 1). There were no false negatives reported. The incorporation of a second-tier genetic screening test by NGS greatly enhanced our program's performance with 5-working days turn-around time maintained as before. In addition, early genetic information is available at the time of recall to facilitate better clinical management and genetic counseling.
Publicações recentes
Structural basis of sodium ion-dependent carnitine transport by OCTN2.
Clinical, Biochemical and Molecular Characterisation of Newborns With Fatty Acid β-Oxidation Disorders: Novel Variants in the ACADM , ACADVL and SLC22A5 Genes.
Systemic Primary Carnitine Deficiency Presenting With Substantia Nigra and Basal Ganglia Injury: A Case Report.
Primary Carnitine Deficiency.
The Human OCTN Sub-Family: Gene and Protein Structure, Expression, and Regulation.
📚 EuropePMC12 artigos no totalmostrando 23
Structural basis of sodium ion-dependent carnitine transport by OCTN2.
Nature communicationsClinical, Biochemical and Molecular Characterisation of Newborns With Fatty Acid β-Oxidation Disorders: Novel Variants in the ACADM , ACADVL and SLC22A5 Genes.
Clinical geneticsSystemic Primary Carnitine Deficiency Presenting With Substantia Nigra and Basal Ganglia Injury: A Case Report.
JIMD reportsThe Human OCTN Sub-Family: Gene and Protein Structure, Expression, and Regulation.
International journal of molecular sciencesHarnessing Next-Generation Sequencing as a Timely and Accurate Second-Tier Screening Test for Newborn Screening of Inborn Errors of Metabolism.
International journal of neonatal screeningSystemic primary carnitine deficiency induces severe arrhythmia due to shortening of QT interval.
Molecular genetics and metabolismA Retrospective Analysis of Clinically Focused Exome Sequencing Results of 372 Infants with Suspected Monogenic Disorders in China.
Pharmacogenomics and personalized medicineRare case of primary carnitine deficiency presenting as acute liver failure.
BMJ case reportsSystemic Primary Carnitine Deficiency: A Case Report with Homozygoys SLC22A5 Gene Mutation.
Klinische PadiatrieDiagnosis of inborn errors of metabolism within the expanded newborn screening in the Madrid region.
JIMD reportsNewborn screening for carnitine transporter defect in Bavaria and the long-term follow-up of the identified newborns and mothers: Assessing the benefit and possible harm based on 19 ½ years of experience.
Molecular genetics and metabolism reportsTargeted Therapies for Metabolic Myopathies Related to Glycogen Storage and Lipid Metabolism: a Systematic Review and Steps Towards a 'Treatabolome'.
Journal of neuromuscular diseasesFirst Case Report of Primary Carnitine Deficiency Manifested as Intellectual Disability and Autism Spectrum Disorder.
Brain sciencesPrimary carnitine deficiency with severe acute hepatitis following rotavirus gastroenteritis.
Journal of infection and chemotherapy : official journal of the Japan Society of ChemotherapyA newborn with seizures born to a mother diagnosed with primary carnitine deficiency.
BMC pediatricsA case of atypical systemic primary carnitine deficiency in Saudi Arabia.
Pediatric reportsPrimary carnitine deficiency in a 57-year-old patient with recurrent exertional rhabdomyolysis.
BMJ case reportsSLC22A5 Mutations in a Patient With Systemic Primary Carnitine Deficiency and Cleft Palate-Successful Perioperative Management.
The Journal of craniofacial surgeryBiochemical characteristics of newborns with carnitine transporter defect identified by newborn screening in California.
Molecular genetics and metabolism[Mutational analysis of SLC22A5 gene in eight patients with systemic primary carnitine deficiency].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsSystemic primary carnitine deficiency with hypoglycemic encephalopathy.
Annals of pediatric endocrinology & metabolismPrimary Carnitine Deficiency - A Rare Treatable Cause of Cardiomyopathy and Massive Hepatomegaly.
Indian journal of pediatricsPrimary systemic carnitine deficiency: a Turkish case with a novel homozygous SLC22A5 mutation and 14 years follow-up.
Journal of pediatric endocrinology & metabolism : JPEMAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Deficiência de carnitina primária sistêmica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Deficiência de carnitina primária sistêmica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Clinical, Biochemical and Molecular Characterisation of Newborns With Fatty Acid β-Oxidation Disorders: Novel Variants in the ACADM , ACADVL and SLC22A5 Genes.
- Structural basis of sodium ion-dependent carnitine transport by OCTN2.
- Systemic Primary Carnitine Deficiency Presenting With Substantia Nigra and Basal Ganglia Injury: A Case Report.
- The Human OCTN Sub-Family: Gene and Protein Structure, Expression, and Regulation.
- Harnessing Next-Generation Sequencing as a Timely and Accurate Second-Tier Screening Test for Newborn Screening of Inborn Errors of Metabolism.
- Primary Carnitine Deficiency.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:158(Orphanet)
- OMIM OMIM:212140(OMIM)
- MONDO:0008919(MONDO)
- GARD:5104(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q3358135(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
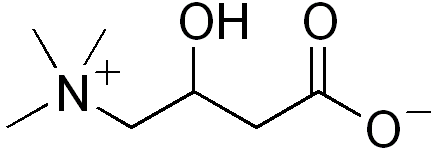
Deficiência de carnitina primária sistêmica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov