A síndrome de Jeune, também chamada de distrofia torácica asfixiante, é uma displasia de costelas curtas caracterizada por tórax estreito, membros curtos e anormalidades esqueléticas radiológicas, incluindo aspecto de "tridente" do acetábulo e alterações metafisárias.
Introdução
O que você precisa saber de cara
A síndrome de Jeune, também chamada de distrofia torácica asfixiante, é uma displasia de costelas curtas caracterizada por tórax estreito, membros curtos e anormalidades esqueléticas radiológicas, incluindo aspecto de "tridente" do acetábulo e alterações metafisárias.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 159 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 388 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
25 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
May function as a motor for intraflagellar retrograde transport. Functions in cilia biogenesis. May play a role in transport between endoplasmic reticulum and Golgi or organization of the Golgi in cells (By similarity)
Cytoplasm, cytoskeleton, cilium axonemeCell membraneCytoplasm
Short-rib thoracic dysplasia 3 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Phosphorylates a large number of substrates in the cytoplasm and the nucleus (PubMed:15642694, PubMed:15905176, PubMed:16387847, PubMed:17333334, PubMed:17565987, PubMed:17693412, PubMed:18836454, PubMed:19949837, PubMed:20356841, PubMed:21085490, PubMed:21514275, PubMed:21812984, PubMed:21852232, PubMed:31112131). Phosphorylates CDC25B, ABL1, NFKB1, CLDN3, histone H1.4 (H1-4), PSMC5/RPT6, PJA2, RYR2, RORA, SOX9, UHRF1 and VASP (PubMed:15178447, PubMed:15642694, PubMed:15905176, PubMed:16387847,
CytoplasmCell membraneMembraneNucleusMitochondrionCell projection, cilium, flagellumCytoplasmic vesicle, secretory vesicle, acrosome
Primary pigmented nodular adrenocortical disease 4
A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Adrenal glands show overall normal size and weight, and multiple small yellow-to-dark brown nodules surrounded by a cortex with a uniform appearance. Microscopically, there are moderate diffuse cortical hyperplasia with mostly nonpigmented nodules, multiple capsular deficits and massive circumscribed and infiltrating extra-adrenal cortical excrescences with micronodules. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes.
Mediates cAMP-dependent signaling triggered by receptor binding to GPCRs (PubMed:12420224, PubMed:21423175, PubMed:31112131). PKA activation regulates diverse cellular processes such as cell proliferation, the cell cycle, differentiation and regulation of microtubule dynamics, chromatin condensation and decondensation, nuclear envelope disassembly and reassembly, as well as regulation of intracellular transport mechanisms and ion flux (PubMed:12420224, PubMed:21423175). Regulates the abundance o
CytoplasmCell membraneMembraneNucleus
Cardioacrofacial dysplasia 2
An autosomal dominant disease characterized by dysmorphic facial features, congenital cardiac defects, primarily common atrium or atrioventricular septal defect, and limb anomalies, including short limbs, brachydactyly and postaxial polydactyly. CAFD2 patients may show developmental delay of variable severity, intellectual disability, autistic features and focal seizures.
As a component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is required in ciliogenesis and ciliary protein trafficking (PubMed:27932497, PubMed:29220510). Involved in cilia formation during neuronal patterning. Acts as a negative regulator of Shh signaling. Required to recruit TULP3 to primary cilia (By similarity)
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Cranioectodermal dysplasia 1
A disorder characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include dolichocephaly (with or without sagittal suture synostosis), scaphocephaly, short stature, limb shortening, short ribs, narrow chest, brachydactyly, renal failure and hepatic fibrosis, small and abnormally shaped teeth, sparse hair, skin laxity and abnormal nails.
Acts as a transcriptional activator (PubMed:10806483, PubMed:19706761, PubMed:19878745, PubMed:24076122, PubMed:24217340, PubMed:24311597). Binds to the DNA consensus sequence 5'-GACCACCCA-3' (PubMed:2105456, PubMed:24217340, PubMed:8378770). Regulates the transcription of specific genes during normal development (PubMed:19706761). Plays a role in craniofacial development and digital development, as well as development of the central nervous system and gastrointestinal tract. Mediates SHH signal
CytoplasmNucleus
Polydactyly, postaxial, A8
A form of postaxial polydactyly, a condition characterized by the occurrence of supernumerary digits in the upper and/or lower extremities. In postaxial polydactyly type A, the extra digit is well-formed and articulates with the fifth or a sixth metacarpal/metatarsal. PAPA8 is an autosomal recessive condition characterized by the presence of postaxial extra digits (hexadactyly) on the hands and/or the feet.
As a component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in ciliogenesis and ciliary protein trafficking (PubMed:21473986, PubMed:28400947, PubMed:29220510). May promote CASP3 activation and TNF-stimulated apoptosis
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, cilium basal body
Cranioectodermal dysplasia 2
A disorder characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include short stature, dolichocephaly, craniosynostosis, narrow thorax with pectus excavatum, short limbs, brachydactyly, joint laxity, narrow palpebral fissures, telecanthus with hypertelorism, low-set simple ears, everted lower lip, and short neck. Teeth abnormalities include widely spaced, hypoplastic and fused teeth.
Phosphorylates serines and threonines, but also appears to possess tyrosine kinase activity (PubMed:20230784). Involved in DNA damage checkpoint control and for proper DNA damage repair (PubMed:20230784). In response to injury that includes DNA damage, NEK1 phosphorylates VDAC1 to limit mitochondrial cell death (PubMed:20230784). May be implicated in the control of meiosis (By similarity). Involved in cilium assembly (PubMed:21211617)
NucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm
Short-rib thoracic dysplasia 6 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
As a component of IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in ciliogenesis (PubMed:28400947, PubMed:28973684). Involved in retrograde ciliary transport along microtubules from the ciliary tip to the base (PubMed:21378380)
Cytoplasm, cytoskeletonCell projection, cilium
Cranioectodermal dysplasia 3
A disorder primarily characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include craniosynostosis, narrow rib cage, short limbs, brachydactyly, hypoplastic and widely spaced teeth, sparse hair, skin laxity and abnormal nails. Nephronophthisis leading to progressive renal failure, hepatic fibrosis, heart defects, and retinitis pigmentosa have also been described.
Plays a key role in ciliogenesis and embryonic development. Regulator of cilia formation by controlling the organization of the apical actin cytoskeleton and the positioning of the basal bodies at the apical cell surface, which in turn is essential for the normal orientation of elongating ciliary microtubules. Plays a key role in definition of cell polarity via its role in ciliogenesis but not via conversion extension. Has an indirect effect on hedgehog signaling (By similarity). Proposed to fun
CytoplasmCell surfaceCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Short-rib thoracic dysplasia 20 with polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Involved in ciliogenesis as part of a complex involved in intraflagellar transport (IFT), the bi-directional movement of particles required for the assembly, maintenance and functioning of primary cilia (PubMed:27466190). Required for the anterograde transport of IFT88 (PubMed:27466190)
Cell projection, cilium
Short-rib thoracic dysplasia 16 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Required for ciliogenesis and sonic hedgehog/SHH signaling. Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1. May play a role in early ciliogenesis in the disappearance of centriolar satellites that preceeds ciliary vesicle formation (PubMed:24421332). Involved in regulation of cell intracellular organization. Involved in regulation of cell polarity (By similarity). Required for asymmetrical localization of CEP
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomePhotoreceptor inner segmentCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 23
A mild form of Joubert syndrome, a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Component of the intraflagellar transport (IFT) complex B: together with IFT74, forms a tubulin-binding module that specifically mediates transport of tubulin within the cilium. Binds tubulin via its CH (calponin-homology)-like region (PubMed:23990561). Required for ciliogenesis (PubMed:23990561, PubMed:27666822). Required for proper regulation of SHH signaling (PubMed:27666822). Plays an important role during spermatogenesis by modulating the assembly and elongation of the sperm flagella (By si
Cell projection, ciliumCytoplasmCytoplasm, cytoskeleton, cilium basal body
Short-rib thoracic dysplasia 19 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system. Required for proper retrograde ciliary transport
Dynein axonemal particle
Short-rib thoracic dysplasia 17 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Involved in endochondral growth and skeletal development
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membrane
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Plays a critical role in bone formation and skeletal development. May be involved in early embryonic morphogenesis
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membraneNucleus
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Required for the maintenance and formation of cilia. Plays an indirect role in hedgehog (Hh) signaling, cilia being required for all activity of the hedgehog pathway (By similarity)
Cell projection, cilium
Short-rib thoracic dysplasia 10 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
As component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in cilia function and/or assembly (PubMed:20889716). Essential for functional IFT-A assembly and ciliary entry of GPCRs (PubMed:20889716). Associates with the BBSome complex to mediate ciliary transport (By similarity)
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCell projection, cilium, photoreceptor outer segmentCell projection, cilium, flagellum
Cranioectodermal dysplasia 4
A disorder primarily characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include craniosynostosis, narrow rib cage, short limbs, brachydactyly, hypoplastic and widely spaced teeth, sparse hair, skin laxity and abnormal nails. Nephronophthisis leading to progressive renal failure, hepatic fibrosis, heart defects, and retinitis pigmentosa have also been described.
Component of the intraflagellar transport (IFT) complex B, which is essential for the development and maintenance of motile and sensory cilia
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axoneme
Short-rib thoracic dysplasia 2 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Involved in centriole duplication (PubMed:24613305, PubMed:26297806). Positively regulates CEP63 centrosomal localization (PubMed:24613305, PubMed:26297806). Required for WDR62 centrosomal localization and promotes the centrosomal localization of CDK2 (PubMed:24613305, PubMed:26297806). May play a role in cilium assembly
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satelliteCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Orofaciodigital syndrome 15
A form of orofaciodigital syndrome, a group of heterogeneous disorders characterized by malformations of the oral cavity, face and digits, and associated phenotypic abnormalities that lead to the delineation of various subtypes. OFD15 features include facial dysmorphism, lobulated tongue, clefting of the alveolar ridges, left hand postaxial polydactyly, broad right hallux and left hallux duplication, and intermittent respiratory difficulty. Brain anomalies include vermis hypoplasia with molar tooth sign, agenesis of corpus callosum, and ventricular dilation. OFD15 inheritance is autosomal recessive.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system (PubMed:23910462, PubMed:25205765, PubMed:29742051, PubMed:31451806). DYNC2I1 plays a major role in retrograde ciliary protein trafficking in cilia and flagella (PubMed:29742051, PubMed:30320547, PubMed:30649997). Also requi
Cell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 8 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system, facilitating the assembly of these organelles (PubMed:29742051). Involved in the regulation of ciliary length (PubMed:26077881, PubMed:26130459)
Golgi apparatusCytoplasmCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 15 with polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs). Essential for retrograde trafficking of IFT-1, IFT-B and GPCRs (PubMed:27932497). Negatively modulates the SHH signal transduction (By similarity)
Cytoplasm, cytoskeleton, cilium axoneme
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system (PubMed:25205765, PubMed:29742051). DYNC2I2 plays a major role in retrograde ciliary protein trafficking and in ciliogenesis (PubMed:29742051, PubMed:30320547, PubMed:30649997). Required also to maintain a functional transit
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, ciliumCell projection, filopodium
Short-rib thoracic dysplasia 11 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs) (PubMed:20889716, PubMed:22503633). Plays a pivotal role in proper development and function of ciliated cells through its role in ciliogenesis and/or cilium maintenance (PubMed:22503633). Required for the development and maintenance of the outer segments of rod and cone photoreceptor cells. Plays a role in maintenance and the delivery of opsin to
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, cilium
Short-rib thoracic dysplasia 9 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome. SRTD9 is characterized by phalangeal cone-shaped epiphyses, chronic renal disease, nearly constant retinal dystrophy, and mild radiographic abnormality of the proximal femur. Occasional features include short stature, cerebellar ataxia, and hepatic fibrosis.
Plays a role in the microtubule-dependent coupling of the nucleus and the centrosome. Involved in the processes that regulate centrosome-mediated interkinetic nuclear migration (INM) of neural progenitors and for proper positioning of neurons during brain development. Also implicated in the migration and selfrenewal of neural progenitors. Required for centriole duplication and maturation during mitosis and subsequent ciliogenesis (By similarity). Required for the recruitment of CEP295 to the pro
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 13 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Variantes genéticas (ClinVar)
911 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
41 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Jeune
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
A Letter from the Mother of a Child with Jeune Syndrome: A Qualitative Analysis.
This study explored a mother's experience of caring for her child with Jeune syndrome through a phenomenological qualitative approach using letter writing. Data were analyzed inductively, and four major themes emerged: challenges and successes in medical treatment, emotional resilience and family dynamics, overcoming developmental and educational barriers, and hopes and aspirations for the future. The mother described interactions with health care providers, her child's surgeries, treatments, and postoperative care. She emphasized the importance of family support, emotional struggles, and concerns about her child's future health. Developmental delays, social difficulties, and limited access to formal education were noted. Future plans involved tracheostomy closure, dietary improvements, and genetic testing. Despite these challenges, the mother remained hopeful, emphasizing her child's postoperative recovery and potential for greater independence. This analysis highlights the struggles, victories, and aspirations of a mother facing a rare disease and offers inspiration to others in similar circumstances.
Retinal astrocytoma and Jeune syndrome relationship from ciliopathy perspective: a case report.
Jeune syndrome is an autosomal recessive chondrodysplasia characterized by skeletal deformities and extra-skeletal organ involvement. Retinal astrocytic hamartomas (astrocytomas) are benign glial cell 10 15 Q1 tumors that are generally asymptomatic and diagnosed incidentally. The IFT74 gene is responsible for the formation of IFT proteins, which play a major role in ciliogenesis. In this retrospective clinical laboratory observational study, an 18-year-old male with Jeune syndrome and night vision loss is presented. Fundus examination revealed bilateral optic discs with minimally blurred margins and bilateral retinal pigment epithelium changes in salt-pepper pattern in the peripheral retina. Additionally, a yellowish retinal astrocytoma was observed inferior to the optic disc in the left eye. Genetic analysis of the patient revealed a homozygous deletion in exon 2 of the IFT74 gene. Our observations on this patient and some relationships between hamartoma and ciliopathies other than eye may potentially suggest a possible association between Jeune syndrome and retinal astrocytoma in the context of IFT74-related ciliopathies.
Ciliopathy-Associated Missense Mutations in IFT140 are Tolerated by the Inherent Resilience of the IFT Machinery.
Genotype-phenotype correlations of rare diseases are complicated by low patient number, high phenotype variability, and compound heterozygosity. Mutations may cause instability of single proteins, and affect protein complex formation or overall robustness of a specific process in a given cell. Ciliopathies offer an interesting case for studying genotype-phenotype correlations as they have a spectrum of severity and include diverse phenotypes depending on different mutations in the same protein. For instance, mutations in the intraflagellar transport protein IFT140 cause a vast spectrum of ciliopathies ranging from isolated retinal dystrophy to severe skeletal abnormalities and multi-organ diseases such as Mainzer-Saldino and Jeune syndrome. Here, the quantitative effects of 23 missense mutations in IFT140, which forms part of the crucial IFT-A complex of the ciliary machinery, were analyzed using affinity purification coupled with mass spectrometry (AP-MS). A subset of 10 mutations led to a significant and domain-specific reduction in IFT140-IFT-A complex interaction indicating complex formation issues and potentially hampering its molecular function. Knockout of IFT140 led to loss of cilia, as shown before. However, phenotypically only mild effects concerning cilia assembly were observed for two out of four tested IFT140 missense mutations. Therefore, our results demonstrate the utility of AP-MS in discerning pathogenic MMs from polymorphisms, and we postulate that reduced function is tolerated by the evolutionarily highly conserved IFT-A system.
Secondary Asphyxiating Thoracic Dysplasia Due to Multiple Chondromas: A Novel Surgical Report.
Asphyxiating thoracic dysplasia (ATD), also known as Jeune syndrome, is a rare and serious genetic condition; its incidence in adult populations is even rarer. A 25-year-old male had a 10-year history of chest wall deformity and progressive dyspnoea. A complex chest wall reconstruction, along with the excision of bone tumours, was performed in view of critical hypoxia. Mechanical ventilation was persistently required postoperatively. However, the patient did improve, and eventually proper chest configuration was restored with a special surgical technique. Histopathological analysis demonstrated the presence of multiple osteochondromas of the ribs. To the best of our knowledge, this is the first reported case of secondary ATD caused by osteochondromas of the ribs.
Enhancing rare disease detection with deep phenotyping from EHR narratives: evaluation on Jeune syndrome.
Patients with rare diseases frequently experience misdiagnoses and long diagnostic delays. Accelerating their diagnosis is essential to ensure timely access to appropriate care. Given the increasing availability of EHRs, combining artificial intelligence and deep phenotyping from large-scale clinical databases offers a promising approach to identify undiagnosed patients. This study assesses the impact of improved phenotype extraction on a screening algorithm for Jeune syndrome, a rare ciliopathy characterized by skeletal abnormalities. Phenotypes from Jeune syndrome patients and controls were automatically extracted from patient unstructured EHRs relying on two thesauri separately: the standard UMLS Metathesaurus and the UMLS+, an enhanced version incorporating additional terms identified through deep learning. The machine learning pipeline that we designed for classifying patients with renal ciliopathy was adapted for Jeune syndrome detection. The model was trained and tested on both the datasets created using the two phenotyping strategies. Using UMLS+ strongly improved the classification of patients with Jeune syndrome, increasing the sensitivity from 49 % to 95 % while maintaining a 90 % specificity. The review of a subset of misclassified controls showed that most of them (69 %) had other genetic skeletal disorders, indicating that the model also captured patients who would benefit from referral to a bone disease geneticist. AI-based screening combined with high-quality deep phenotyping can help reduce diagnostic delay in rare diseases. The completeness and accuracy of phenotyping from EHRs have a strong impact on screening performances.
Publicações recentes
A Letter from the Mother of a Child with Jeune Syndrome: A Qualitative Analysis.
Secondary Asphyxiating Thoracic Dysplasia Due to Multiple Chondromas: A Novel Surgical Report.
Retinal astrocytoma and Jeune syndrome relationship from ciliopathy perspective: a case report.
🥉 Relato de casoEnhancing rare disease detection with deep phenotyping from EHR narratives: evaluation on Jeune syndrome.
Phenotypic heterogeneity in DYNC2H1-related short-rib thoracic dysplasia: antenatal indicators and postnatal outcomes.
📚 EuropePMC102 artigos no totalmostrando 89
A Letter from the Mother of a Child with Jeune Syndrome: A Qualitative Analysis.
Clinical pediatricsSecondary Asphyxiating Thoracic Dysplasia Due to Multiple Chondromas: A Novel Surgical Report.
Interdisciplinary cardiovascular and thoracic surgeryRetinal astrocytoma and Jeune syndrome relationship from ciliopathy perspective: a case report.
Ophthalmic geneticsEnhancing rare disease detection with deep phenotyping from EHR narratives: evaluation on Jeune syndrome.
International journal of medical informaticsPhenotypic heterogeneity in DYNC2H1-related short-rib thoracic dysplasia: antenatal indicators and postnatal outcomes.
Journal of medical geneticsReport of a Rare Syndromic Retinal Dystrophy: Asphyxiating Thoracic Dystrophy (Jeune Syndrome).
Turkish journal of ophthalmologyA novel compound heterozygous mutation in the DYNC2H1 gene in a Chinese family with Jeune syndrome.
HereditasCiliopathy-Associated Missense Mutations in IFT140 are Tolerated by the Inherent Resilience of the IFT Machinery.
Molecular & cellular proteomics : MCPCoding and non-coding variants in the ciliopathy gene CFAP410 cause early-onset non-syndromic retinal degeneration.
NPJ genomic medicineEarly-onset renal dysfunction in Jeune syndrome: A case report with atypical presentation.
Radiology case reportsEpithelial Predominant Wilms Tumor in an Adult Patient: Case Report and Literature Review.
Journal of kidney cancer and VHLA novel GRK2 variant in a patient with Jeune asphyxiating thoracic dysplasia accompanied by Morgagni hernia.
American journal of medical genetics. Part AReanalysis of Whole-Exome Sequencing Data of an Infant with Suspected Diagnosis of Jeune Syndrome Revealed a Likely Pathogenic Variant in GRK2: A Newly Associated Gene for Jeune Syndrome Phenotype.
Molecular syndromologyWhole-Exome Sequencing Identifies DYNC2H1 Mutations as a Cause of Jeune Asphyxiating Thoracic Dystrophy Without Extra-Skeletal Organ Involvement.
International medical case reports journalA Mild Case of Jeune Syndrome Associated with a Recurrent Missense Variant in DYNC2H1: Confirmation of a Genotype-Phenotype Correlation.
Klinische PadiatrieCompound heterozygous WDR19 variants associated with nephronophthisis, Caroli disease, refractory epilepsy and congenital bilateral central blindness: Case report.
HeliyonClinical features and genetic analysis of a case series of skeletal ciliopathies in a prenatal setting.
BMC medical genomicsCase report: A simple and reliable approach for progressive internal distraction of the sternum for Jeune syndrome (asphyxiating thoracic dystrophy): preliminary experience and literature review of surgical techniques.
Frontiers in pediatricsClinical variability in DYNC2H1-related skeletal ciliopathies includes Ellis-van Creveld syndrome.
European journal of human genetics : EJHGLateral Thoracic Expansion for Jeune's Syndrome, Surgical Approach, and Technical Details.
European journal of pediatric surgery : official journal of Austrian Association of Pediatric Surgery ... [et al] = Zeitschrift fur KinderchirurgieDisease-associated mutations in WDR34 lead to diverse impacts on the assembly and function of dynein-2.
Journal of cell scienceMeier-Gorlin Syndrome: Clinical Misdiagnosis, Genetic Testing and Functional Analysis of ORC6 Mutations and the Development of a Prenatal Test.
International journal of molecular sciencesNPHP3 splice acceptor site variant is associated with infantile nephronophthisis and asphyxiating thoracic dystrophy; A rare combination.
European journal of medical geneticsPrenatal Diagnosis of Jeune Syndrome Caused by Compound Heterozygous Variants in DYNC2H1 Gene-Case Report with Rapid WES Procedure and Differential Diagnosis of Lethal Skeletal Dysplasias.
GenesEnriching UMLS-Based Phenotyping of Rare Diseases Using Deep-Learning: Evaluation on Jeune Syndrome.
Studies in health technology and informaticsThoracic insufficiency syndrome: Approaches to assessment and management.
Paediatric respiratory reviewsSurgical treatment of a 36-year-old patient with asphyxiating thoracic dysplasia.
Interactive cardiovascular and thoracic surgery[Chest Wall Deformities in Children and Adolescents].
Zentralblatt fur ChirurgieTHE CASE OF JEUNE SYNDROME AMONG THE PRECARPATHIAN POPULATION.
Wiadomosci lekarskie (Warsaw, Poland : 1960)A curious case of asphyxiating thoracic dystrophy in an adult.
Respirology case reportsRole of Primary Cilia in Bone and Cartilage.
Journal of dental researchFetal ciliopathies: a retrospective observational single-center study.
Archives of gynecology and obstetricsTtc30a affects tubulin modifications in a model for ciliary chondrodysplasia with polycystic kidney disease.
Proceedings of the National Academy of Sciences of the United States of AmericaClinical phenocopies of albinism.
Journal of AAPOS : the official publication of the American Association for Pediatric Ophthalmology and StrabismusA new technique for neonatal Jeune syndrome: External thoracic expansion.
Turk gogus kalp damar cerrahisi dergisiClinical exome sequencing facilitates the understanding of genetic heterogeneity in Leber congenital amaurosis patients with variable phenotype in southern India.
Eye and vision (London, England)Overlapping holoprosencephaly-polydactyl syndrome and asphyxiating thoracic dystrophy, an incidental finding in late prenatal ultrasound: A rare case report.
Clinical case reportsThirteen Years' Progression of Macular Atrophy in a Patient With Jeune Syndrome.
Ophthalmic surgery, lasers & imaging retinaFirst Report of Spinal Anesthesia for Cesarean Delivery in a Parturient With Jeune Syndrome: A Case Report.
A&A practiceBiallelic loss of function variant in the unfolded protein response gene PDIA6 is associated with asphyxiating thoracic dystrophy and neonatal-onset diabetes.
Clinical geneticsExpression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development.
BMC developmental biologyMutations in GRK2 cause Jeune syndrome by impairing Hedgehog and canonical Wnt signaling.
EMBO molecular medicineOral rehabilitation in a patient with Jeune syndrome presenting with multiple teeth agenesis.
Special care in dentistry : official publication of the American Association of Hospital Dentists, the Academy of Dentistry for the Handicapped, and the American Society for Geriatric DentistryFetal micromelia, thoracic dysplasia and polydactyly revisited: A case-based antenatal sonographic approach.
Ultrasound (Leeds, England)Prophylactic Decompression for Cervical Stenosis in Jeune Syndrome: Report From a Single Institution.
SpinePrenatal Diagnosis of Jeune Syndrome by Whole-Exome Sequencing in a Case With Mild Skeletal Changes.
Journal of ultrasound in medicine : official journal of the American Institute of Ultrasound in MedicineRETINAL DYSTROPHY IN JEUNE SYNDROME: A MULTIMODAL IMAGING CHARACTERIZATION.
Retinal cases & brief reportsAttenuated Type of Asphyxiating Thoracic Dysplasia due to Mutations in DYNC2H1 Gene.
Prague medical reportA new case of KIAA0753-related variant of Jeune asphyxiating thoracic dystrophy.
European journal of medical geneticsFetal magnetic resonance imaging of skeletal dysplasias.
Pediatric radiology[Bilateral papilledema in a thirteen-year-old girl with Jeune syndrome].
Journal francais d'ophtalmologieCEP120 interacts with C2CD3 and Talpid3 and is required for centriole appendage assembly and ciliogenesis.
Scientific reportsDown-regulated WDR35 contributes to fetal anomaly via regulation of osteogenic differentiation.
GeneA global analysis of IFT-A function reveals specialization for transport of membrane-associated proteins into cilia.
Journal of cell scienceAn unusual case of hypercapnic respiratory failure.
Respiratory medicine case reportsDynein-2 intermediate chains play crucial but distinct roles in primary cilia formation and function.
eLifeAnesthetic management of an infant with Jeune syndrome and severe pulmonary hypertension for tracheostomy.
Journal of clinical anesthesiaAlu-Alu mediated intragenic duplications in IFT81 and MATN3 are associated with skeletal dysplasias.
Human mutationExtracorporeal membrane oxygenation support in individuals with thoracic insufficiency.
PerfusionPrimary presentation of Jeune's syndrome as gastric motility disorder in an infant: A case report.
The Indian journal of radiology & imagingExpanding the phenotype associated with biallelic WDR60 mutations: Siblings with retinal degeneration and polydactyly lacking other features of short rib thoracic dystrophies.
American journal of medical genetics. Part AExome sequencing for the differential diagnosis of ciliary chondrodysplasias: Example of a WDR35 mutation case and review of the literature.
European journal of medical geneticsBiallelic mutations in DYNC2LI1 are a rare cause of Ellis-van Creveld syndrome.
Clinical geneticsVariable expressivity of TCTEX1D2 mutations and a possible pathogenic link of molar-incisor malformation to ciliary dysfunction.
Archives of oral biologyHomozygous variant in C21orf2 in a case of Jeune syndrome with severe thoracic involvement: Extending the phenotypic spectrum.
American journal of medical genetics. Part ADYNC2H1 mutation causes Jeune syndrome and recurrent lung infections associated with ciliopathy.
The clinical respiratory journalAnesthetic Management of a Child With Jeune Syndrome for Tracheotomy: A Case Report.
A & A case reportsEffective Neurally Adjusted Ventilatory Assist (NAVA) Ventilation in a Child With Jeune Syndrome.
PediatricsMutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type.
Clinical genetics[Infant respiratory distress revealing Jeune syndrome].
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieCompound heterozygous mutations in the IFT140 gene cause Opitz trigonocephaly C syndrome in a patient with typical features of a ciliopathy.
Clinical geneticsDestabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a Spectrum of Short-Rib Polydactyly Syndrome.
Scientific reportsHypertrophic cardiomyopathy with Jeune syndrome: The first reported case.
Turk Kardiyoloji Dernegi arsivi : Turk Kardiyoloji Derneginin yayin organidirSpinal correction of scoliosis in Jeune syndrome: a report of two cases.
Scoliosis and spinal disordersDistraction osteogenesis of the sternum for thoracic expansion in a severe case of jeune syndrome: a preliminary report.
Journal of plastic surgery and hand surgeryAxial Spondylometaphyseal Dysplasia Is Caused by C21orf2 Mutations.
PloS oneNew mutations in DYNC2H1 and WDR60 genes revealed by whole-exome sequencing in two unrelated Sardinian families with Jeune asphyxiating thoracic dystrophy.
Clinica chimica acta; international journal of clinical chemistryThoracic Insufficiency Syndrome.
Current problems in pediatric and adolescent health careRespiratory sleep disorders in Jeune syndrome: a case description.
Archives italiennes de biologieA relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35.
American journal of medical genetics. Part A[Clinical, radiological and auxologic long-term evolution of 8 children with asphyxiating thoracic dysplasia].
Archivos argentinos de pediatriaMutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes.
Journal of medical geneticsAnesthetic Approach for a Patient with Jeune Syndrome.
Case reports in anesthesiologyAn siRNA-based functional genomics screen for the identification of regulators of ciliogenesis and ciliopathy genes.
Nature cell biologyAntenatal Diagnosis of Jeune Syndrome (Asphyxiating Thoracic Dysplasia) with Micromelia and Facial Dysmorphism on Second-Trimester Ultrasound.
Polish journal of radiologyTCTEX1D2 mutations underlie Jeune asphyxiating thoracic dystrophy with impaired retrograde intraflagellar transport.
Nature communicationsRespiratory motile cilia dysfunction in a patient with cranioectodermal dysplasia.
American journal of medical genetics. Part A[Clinical case of Jeune syndrome].
Vestnik rentgenologii i radiologiiManagement of Thoracic Insufficiency Syndrome in Patients With Jeune Syndrome Using the 70 mm Radius Vertical Expandable Prosthetic Titanium Rib.
Journal of pediatric orthopedicsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Jeune.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Jeune
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A Letter from the Mother of a Child with Jeune Syndrome: A Qualitative Analysis.
- Retinal astrocytoma and Jeune syndrome relationship from ciliopathy perspective: a case report.
- Ciliopathy-Associated Missense Mutations in IFT140 are Tolerated by the Inherent Resilience of the IFT Machinery.
- Secondary Asphyxiating Thoracic Dysplasia Due to Multiple Chondromas: A Novel Surgical Report.
- Enhancing rare disease detection with deep phenotyping from EHR narratives: evaluation on Jeune syndrome.
- Phenotypic heterogeneity in DYNC2H1-related short-rib thoracic dysplasia: antenatal indicators and postnatal outcomes.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:474(Orphanet)
- MONDO:0018770(MONDO)
- GARD:3049(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4807981(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
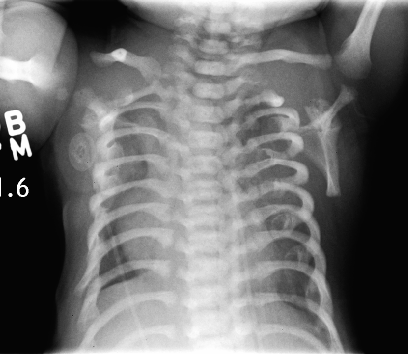
Síndrome Jeune
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata