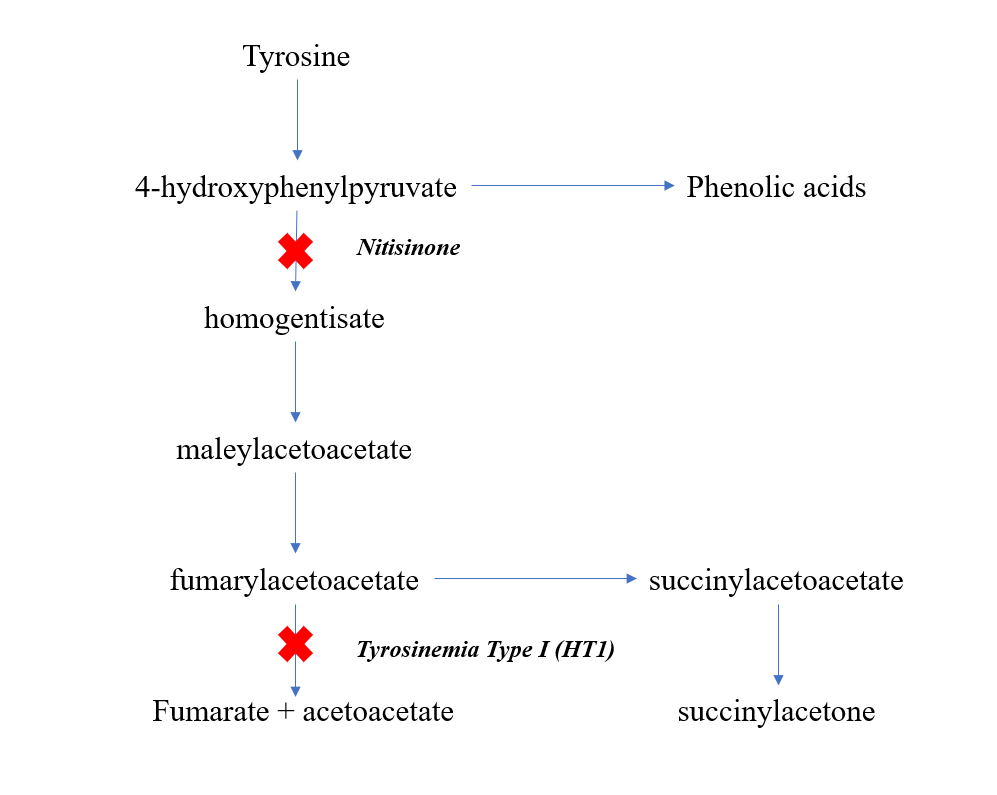
A tirosinemia tipo 1 (HTI) é um erro congênito do catabolismo da tirosina causado por atividade defeituosa da fumarilacetoacetato hidrolase (FAH) e é caracterizada por doença hepática progressiva, disfunção tubular renal, crises semelhantes à porfiria e uma melhora dramática no prognóstico após tratamento com nitisinona.
Introdução
O que você precisa saber de cara
A tirosinemia tipo 1 (HTI) é um erro congênito do catabolismo da tirosina causado por atividade defeituosa da fumarilacetoacetato hidrolase (FAH) e é caracterizada por doença hepática progressiva, disfunção tubular renal, crises semelhantes à porfiria e uma melhora dramática no prognóstico após tratamento com nitisinona.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 14 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 41 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisTyrosinemia 1
An inborn error of metabolism characterized by elevations of tyrosine in the blood and urine, and hepatorenal manifestations. Typical features include hepatic necrosis, renal tubular injury, episodic weakness, self-mutilation, and seizures. Renal tubular dysfunction is associated with phosphate loss and hypophosphataemic rickets. Progressive liver disease can lead to the development of hepatocellular carcinoma. Dietary treatment with restriction of tyrosine and phenylalanine alleviates the rickets, but liver transplantation has so far been the only definite treatment.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
235 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.607 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Tirosinemia tipo 1
Centros de Referência SUS
21 centros habilitados pelo SUS para Tirosinemia tipo 1
Centros para Tirosinemia tipo 1
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
NUPAD / Faculdade de Medicina UFMG
Av. Prof. Alfredo Balena, 189 - 5 andar - Centro, Belo Horizonte - MG, 30130-100 · CNES 2183226
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital de Clínicas da Universidade Federal de Pernambuco
Av. Prof. Moraes Rego, 1235 - Cidade Universitária, Recife - PE, 50670-901 · CNES 2561492
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital Universitário Onofre Lopes (HUOL)
Av. Nilo Peçanha, 620 - Petrópolis, Natal - RN, 59012-300 · CNES 2408570
Atenção Especializada
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Instituto da Criança e do Adolescente (ICr-HCFMUSP)
Av. Dr. Enéas Carvalho de Aguiar, 647 - Cerqueira César, São Paulo - SP, 05403-000 · CNES 2081695
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
21 ensaios clínicos encontrados, 3 ativos.
Publicações mais relevantes
SIADH as an Underrecognized Manifestation of Porphyria-like Crises in Hereditary Tyrosinemia Type 1: Clinical and Pathophysiological Insights.
Hereditary tyrosinemia type 1 (HT1) is a rare metabolic disorder caused by fumarylacetoacetate hydrolase deficiency, leading to the accumulation of toxic metabolites such as fumarylacetoacetate (FAA) and succinylacetone (SA). We report an 11-year-old boy with poorly controlled HT1 who presented with a severe neurovisceral crisis after suboptimal adherence to nitisinone (NTBC) therapy, characterized by abdominal pain, hypertension, paralytic ileus, seizures, and profound hyponatremia. Biochemical evaluation revealed markedly elevated urinary δ-aminolevulinic acid (ALA), consistent with a porphyria-like metabolic decompensation, together with inappropriately increased plasma copeptin in the setting of hypotonic hyponatremia and clinical euvolemia, fulfilling diagnostic criteria for the syndrome of inappropriate antidiuretic hormone secretion (SIADH). Optimization of NTBC therapy combined with tailored fluid management resulted in complete clinical and biochemical recovery. This case supports a pathophysiological link between acute disruption of the heme-porphyrin pathway and inappropriate antidiuretic hormone secretion. In HT1, this susceptibility may be further amplified by FAA- and SA-mediated oxidative stress, mitochondrial dysfunction, and heme depletion, with an additional contribution from SA-associated renal tubular impairment. Overall, our findings underscore SIADH as a potentially underrecognized cause of acute hyponatremia in HT1 and highlight the importance of strict NTBC adherence and early monitoring of urinary ALA during metabolic decompensation.
Identification and Characterization of Novel Variants of Fumarylacetoacetate Hydrolase (FAH) Gene in Clinically Suspected Patients of Tyrosinemia Type 1: Tertiary Care Centre Study of North India.
Tyrosinemia type I is a rare autosomal recessive metabolic disease caused by the deficiency of Fumarylacetoacetate hydrolase (FAH). The deficiency leads to the accumulation of toxic metabolites leading to the hepatorenal complications. The present study was planned for the identification of the spectrum of disease-causing mutations in the FAH gene of North Indian population. 70 clinically suspected tyrosinemia type I patients were recruited. Urinary Succinylacetone was estimated using Gas Chromatography Mass spectrometry (GCMS). The gene FAH was sequenced by Sanger sequencing in the patients whose exons showed band mobility change by single standard conformation polymorphism (SSCP) screening. Identified variants were functionally analyzed using insilico software tools for identification of pathogenic variants with in vitro functional characterization. Urinary Succinylacetone was not detected in urine of the recruited patients. Sequencing analysis revealed 24variants in nine patients. The majority of these variants were predicted to be disease causing by in silico software programs. In vitro, analysis showed that variants L17P + F22I + I373T and G307X, S130C and G307X can reduce protein expression and catalytic activity of FAH. Tyrosinemia type I is a rare disease but has severe mortality and morbidity. In India, diagnostic and treatment strategies are insufficient against this disease; therefore, majority of the cases may remain undiagnosed. Identification of these disease-causing mutations in the recruited study subjects implicate the requirement of neonatal or prenatal screening for Tyrosinemia Type I in future. The online version contains supplementary material available at 10.1007/s12291-024-01236-6.
Quantitative Succinylacetone Measurement by Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) Facilitates Diagnosis, Monitoring, and Characterization of Tyrosinemia Type 1 and Other Hypersuccinylacetonemias.
Tyrosinemia type 1 (HT1), due to deficient activity of fumarylacetoacetate hydrolase, causes accumulation of succinylacetone (SA). SA concentrations in urine and plasma of untreated HT1 patients are typically several thousand-fold higher than normal, hence are readily recognized by traditional diagnostic methods in most cases. However, quantitation of SA in the nanomolar range is important for monitoring patients treated with nitisinone, for identifying attenuated or atypical forms of HT1, and for confirmation or refutation of the diagnosis of HT1 following a positive newborn screen. Our laboratory, a reference centre for diagnosis and monitoring of HT1, previously assayed SA by gas chromatography-mass spectrometry (GC-MS). Three years ago, we upgraded this method by transferring it to a new triple quadrupole technology (GC-MS/MS). A stable isotope dilution process is used, with sample treatment consisting of an oximation step followed by a single liquid-liquid extraction then trimethylsilyl derivatization. Quantitation is based on intensities of the ion transitions m/z 620 → 181 for SA and 625 → 186 for the internal standard. Method validation demonstrated enhanced analytical specificity and sensitivity, with good precision and accuracy. Using GC-MS/MS instead of GC-MS allowed a limit of quantitation of 1 nmol/L while decreasing the required specimen volumes, as well as reducing the number of sample processing steps, chromatographic run time, and instrument maintenance. This assay facilitates laboratory diagnosis and monitoring of HT1, permits identification and characterization of other hypersuccinylacetonemias including maleylacetoacetate isomerase deficiency, and is also a valuable tool for research studies using animal models and cellular models of HT1.
Fusion Peptide-Incorporated Lipid Nanoparticles Boost Endosomal Escape and Enhance Cytosolic mRNA Delivery.
The endosomal escape capability of current mRNA-loaded lipid nanoparticles (mRNA-LNPs) is generally low, which restricts their overall delivery efficiency. To address this limitation, we adopted a strategy inspired by the viral infection mechanism, utilizing fusion peptides to enhance the intracellular release of mRNA. Nine viral-derived and artificial fusion peptides were co-encapsulated within mRNA-LNPs respectively, termed FP-LNPs, and systematically assessed their efficacy in improving mRNA delivery. Notably, the HA2 fusion peptide from the influenza virus demonstrated a marked enhancement in mRNA delivery efficiency both in vitro and in vivo. Under acidic conditions of endosomes, HA2 collaborates with ionizable cationic lipids to facilitate endosomal membrane rupture, thereby promoting the release of mRNA into the cytoplasm and enhancing protein expression. Moreover, the incorporation of fusion peptides into various types of mRNA-LNP formulations significantly improved their in vivo delivery efficiency of mRNA, resulting in improved gene editing outcomes in the liver and lungs. Furthermore, in a Hereditary Tyrosinemia Type 1 (HT-1) mouse model, HA2-LNP significantly boosted FAH protein expression in the liver and more effectively prevented body weight loss and reduced liver fibrosis. Overall, this approach offers a promising strategy for enhancing endosomal escape and boosting the delivery efficiency of mRNA, underscoring its enhanced therapeutic potential.
Management of porphyria-like syndrome in tyrosinemia type 1.
Hereditary tyrosinemia type 1 (HT1) is a rare metabolic disorder that may present with severe hepatic and neurological complications. Acute porphyria-like crises in HT1 are extremely uncommon and may be associated with severe electrolyte disturbances such as hyponatremia due to the syndrome of inappropriate antidiuretic hormone secretion (SIADH). We describe a 9-year-old girl with genetically confirmed HT1 who developed an acute porphyria-like crisis accompanied by severe symptomatic hyponatremia secondary to SIADH. The patient presented with abdominal pain, vomiting, and progressive neurological deterioration culminating in generalized tonic-clonic seizures. Laboratory evaluation revealed profound hyponatremia, elevated urinary succinylacetone, and increased porphyrin precursors. Management included intravenous hemin therapy, fluid restriction, and intensive care support, leading to full neurological recovery without sequelae. To our knowledge, this is the first reported case in Türkiye successfully managed with hemin during an acute porphyria-like episode in HT1 and only the second documented case of SIADH in this context. This case underscores the importance of recognizing SIADH and optimizing fluid management in patients with HT1 presenting with acute neurological crises.
Publicações recentes
Two Years of Expanded Newborn Screening in Russia: High-Throughput Detection of Inherited Metabolic Disorders by Tandem Mass Spectrometry with Next-Generation Sequencing Confirmation.
Identification and Characterization of Novel Variants of Fumarylacetoacetate Hydrolase (FAH) Gene in Clinically Suspected Patients of Tyrosinemia Type 1: Tertiary Care Centre Study of North India.
Quantitative Succinylacetone Measurement by Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) Facilitates Diagnosis, Monitoring, and Characterization of Tyrosinemia Type 1 and Other Hypersuccinylacetonemias.
Fusion Peptide-Incorporated Lipid Nanoparticles Boost Endosomal Escape and Enhance Cytosolic mRNA Delivery.
Management of porphyria-like syndrome in tyrosinemia type 1.
📚 EuropePMC188 artigos no totalmostrando 178
Identification and Characterization of Novel Variants of Fumarylacetoacetate Hydrolase (FAH) Gene in Clinically Suspected Patients of Tyrosinemia Type 1: Tertiary Care Centre Study of North India.
Indian journal of clinical biochemistry : IJCBQuantitative Succinylacetone Measurement by Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) Facilitates Diagnosis, Monitoring, and Characterization of Tyrosinemia Type 1 and Other Hypersuccinylacetonemias.
JIMD reportsFusion Peptide-Incorporated Lipid Nanoparticles Boost Endosomal Escape and Enhance Cytosolic mRNA Delivery.
Advanced materials (Deerfield Beach, Fla.)Management of porphyria-like syndrome in tyrosinemia type 1.
Journal of pediatric endocrinology & metabolism : JPEMOptimizing Peptide Ionizable Lipids Enables Efficient and Low-Toxicity mRNA Delivery for In Vivo Prime Editing and Protein Replacement Therapy.
Advanced materials (Deerfield Beach, Fla.)SIADH as an Underrecognized Manifestation of Porphyria-like Crises in Hereditary Tyrosinemia Type 1: Clinical and Pathophysiological Insights.
International journal of molecular sciencesUntargeted Metabolomics Reveals Metabolic Reprogramming Linked to HCC Risk in Late Diagnosed Tyrosinemia Type 1.
MetabolitesTherapeutic Monitoring of Patients With Hereditary Tyrosinemia Type 1-A Belgian Monocentric Experience.
JIMD reportsIn Vivo Confocal Microscopy and Anterior Segment Optical Coherence Tomography Features of Corneal Pseudodendritic Lesions in Hereditary Tyrosinemia Type 1.
The American journal of case reportsNeurological crisis in tyrosinemia type 1: Essential roles of replacement therapy and nutrition in multidisciplinary management.
Endocrinologia, diabetes y nutricionImproved specificity and efficiency of in vivo adenine base editing therapies with hybrid guide RNAs.
Nature biomedical engineeringVariants in GSTZ1 Gene Underlying Maleylacetoacetate Isomerase Deficiency: Characterization of Two New Individuals and Literature Review.
GenesHereditary Tyrosinemia Type 1: Success and Challenges in Indian Subcontinent.
Indian pediatricsOverview of European Practices for Management of Tyrosinemia Type 1: Towards European Guidelines.
Journal of inherited metabolic diseaseNebulized Lipid Nanoparticles Deliver mRNA to the Liver for Treatment of Metabolic Diseases.
Nano lettersPersistent hyperinsulinemic hypoglycemia of infancy treated at the Hospital Infantil de Especialidades de Chihuahua.
Boletin medico del Hospital Infantil de MexicoNitisinone desensitization protocol, case report of hereditary Tyrosinemia type 1 with successful treatment and outcomes.
Orphanet journal of rare diseasesValidation of Clinical-Grade Electroporation Systems for CRISPR-Cas9-Mediated Gene Therapy in Primary Hepatocytes for the Correction of Inherited Metabolic Liver Disease.
CellsEvaluation of the Performance of Newborn Screening for Tyrosinemia Type 1 in The Netherlands: Suggestions for Improvements Using Additional Biomarkers in Addition to Succinylacetone.
International journal of neonatal screeningIntegrating Machine Learning and Follow-Up Variables to Improve Early Detection of Hepatocellular Carcinoma in Tyrosinemia Type 1: A Multicenter Study.
International journal of molecular sciencesIdentification of the Mutations Spectrum in the Fumarylacetoacetate Hydrolase Gene in Tyrosinemia Type 1 Patients in Northeastern Iran.
Biochemical geneticsAn Unusual Presentation of Tyrosinemia Type 1 in a Pediatric Patient: Case Report and Comprehensive Review.
Clinical case reportsProgress in Gene Therapy for Hereditary Tyrosinemia Type 1.
PharmaceuticsShort and Long-Term Outcomes of Liver Transplantation in Pediatric Patients With Inborn Errors of Metabolism: A Single-Center Study.
Pediatric transplantationHypophosphatemic Rickets as a Key Sign for the Diagnosis of Hereditary Tyrosinemia Type 1: Case Reports and Narrative Review of the Literature.
Revista medica de ChileEvaluation of Neonatal Screening Programs for Tyrosinemia Type 1 Worldwide.
International journal of neonatal screeningOxidative Stress Associated With Increased Reactive Nitrogen Species Generation in the Liver and Kidney Caused by a Major Metabolite Accumulating in Tyrosinemia Type 1.
Cell biochemistry and functionClinical, Biochemical, and Molecular Characteristics of Filipino Patients with Tyrosinemia Type 1.
International journal of neonatal screeningMutation spectrum of Tyrosinemia type I in Iran, A retrospective cohort study.
European journal of medical geneticsIn vivo dissection of the mouse tyrosine catabolic pathway with CRISPR-Cas9 identifies modifier genes affecting hereditary tyrosinemia type 1.
GeneticsA 12-month, longitudinal, intervention study examining a tablet protein substitute preparation in the management of tyrosinemia.
Molecular genetics and metabolism reportsHereditary Tyrosinemia Type-1 With Late Presentation: A Case Report.
CureusConsenso mexicano de tirosinemia tipo 1.
Boletin medico del Hospital Infantil de MexicoModified by the Innovative Drugs and Strategies-Pattern of Selected Indications for Pediatric Liver Transplantation.
Pediatric transplantationHereditary tyrosinaemia type 1 in the absence of succinylacetone: 4-oxo 6-hydroxyhepanoate (4OHHA), a putative diagnostic biomarker.
JIMD reportsEvaluation of surgical strategies and long-term outcomes in pediatric hepatocellular carcinoma.
Pediatric surgery internationalEx vivo gene editing and cell therapy for hereditary tyrosinemia type 1.
Hepatology communicationsDecoding hepatorenal tyrosinemia type 1: Unraveling the impact of early detection, NTBC, and the role of liver transplantation.
Canadian liver journalClinical Spectrum of Hereditary Tyrosinemia Type 1 in a Cohort of Pakistani Children.
Clinical medicine insights. PediatricsA Lithuanian Case of Tyrosinemia Type 1 with a Literature Review: A Rare Cause of Acute Liver Failure in Childhood.
Medicina (Kaunas, Lithuania)A False-Negative Newborn Screen for Tyrosinemia Type 1-Need for Re-Evaluation of Newborn Screening with Succinylacetone.
International journal of neonatal screeningE. coli´s fight against TYROnny: Designing a bacterial strain to tackle tyrosinemia type 1.
Journal of hepatologyElectronic structure and molecular properties of nitisinone and mesotrione in water.
Journal of molecular modelingAn engineered Escherichia coli Nissle strain prevents lethal liver injury in a mouse model of tyrosinemia type 1.
Journal of hepatologyThe effects of phenylalanine and tyrosine levels on dopamine production in rat PC12 cells. Implications for treatment of phenylketonuria, tyrosinemia type 1 and comorbid neurodevelopmental disorders.
Neurochemistry internationalA patient with urinary succinylacetone-negative hereditary tyrosinemia type 1.
Pediatrics international : official journal of the Japan Pediatric SocietyMaleic acid is a biomarker for maleylacetoacetate isomerase deficiency; implications for newborn screening of tyrosinemia type 1.
Journal of inherited metabolic diseaseCase report: ADHD and prognosis in tyrosinemia type 1.
Frontiers in psychiatryShort-term nitisinone discontinuation of hereditary tyrosinemia type 1 mice causes metabolic alterations in glutathione metabolism/biosynthesis and multiple amino acid degradation pathways.
Genes & diseasesApplication of machine learning tools and integrated OMICS for screening and diagnosis of inborn errors of metabolism.
Metabolomics : Official journal of the Metabolomic SocietyHereditary Tyrosinemia Type 1 Mice under Continuous Nitisinone Treatment Display Remnants of an Uncorrected Liver Disease Phenotype.
GenesIdentification and functional characterization of a novel homozygous intronic variant in the fumarylacetoacetate hydrolase gene in a Chinese patient with tyrosinemia type 1.
BMC medical genomicsInitial presentation, management and follow-up data of 33 treated patients with hereditary tyrosinemia type 1 in the absence of newborn screening.
Molecular genetics and metabolism reportsThe compound heterozygous mutations of c.607G>a and c.657delC in the FAH gene are associated with renal damage with hereditary tyrosinemia type 1 (HT1).
Molecular genetics & genomic medicineComprehensive Evaluation of the NeoBase 2 Non-derivatized MSMS Assay and Exploration of Analytes With Significantly Different Concentrations Between Term and Preterm Neonates.
Annals of laboratory medicineActivation of homology-directed DNA repair plays key role in CRISPR-mediated genome correction.
Gene therapyIn vivo lentiviral vector gene therapy to cure hereditary tyrosinemia type 1 and prevent development of precancerous and cancerous lesions.
Nature communicationsmRNA-based therapy proves superior to the standard of care for treating hereditary tyrosinemia 1 in a mouse model.
Molecular therapy. Methods & clinical developmentBiochemical and behavioural profile of NTBC treated Tyrosinemie type 1 mice.
Molecular genetics and metabolismA case report of two siblings with hypertyrosinemia type 1 presenting with hepatic disease with different onset time and severity.
Molecular genetics and metabolism reportsLiver Transplantation for Tyrosinemia Type 1 in the Developing World: Is It Really the Best We Can Offer?: Authors' Reply.
Indian journal of pediatricsNeurocognitive outcome and mental health in children with tyrosinemia type 1 and phenylketonuria: A comparison between two genetic disorders affecting the same metabolic pathway.
Journal of inherited metabolic diseaseLiver Transplantation for Tyrosinemia Type 1 in the Developing World: Is It Really the Best We Can Offer?: Correspondence.
Indian journal of pediatricsHepatocellular carcinoma requiring liver transplantation in hereditary tyrosinemia type 1 despite nitisinone therapy and α1-fetoprotein normalization.
Pediatric transplantationTyrosine catabolites influence SKN-1 signaling in a model of Type I Tyrosinemia.
microPublication biologyClinical and biochemical footprints of inherited metabolic diseases. VIII. Neoplasias.
Molecular genetics and metabolismSimultaneous newborn screening for sickle cell disease, biotinidase deficiency, and hereditary tyrosinemia type 1 with an optimized tandem mass spectrometry protocol.
Annals of hematologyDiagnosis of inborn errors of metabolism within the expanded newborn screening in the Madrid region.
JIMD reportsLiver Transplantation for Tyrosinemia Type 1 in the Developing World: Is It Really the Best We Can Offer?
Indian journal of pediatricsGeneration of immunodeficient pig with hereditary tyrosinemia type 1 and their preliminary application for humanized liver.
Cell & bioscienceClinical and genetic characteristics of two patients with tyrosinemia type 1 in Slovenia - A novel fumarylacetoacetate hydrolase (FAH) intronic disease-causing variant.
Molecular genetics and metabolism reportsDevelopment of Flow Injection Analysis Method for the Second-Tier Estimation of Succinylacetone in Dried Blood Spot of Newborn Screening.
Indian journal of clinical biochemistry : IJCBNTBC Treatment Monitoring in Chilean Patients with Tyrosinemia Type 1 and Its Association with Biochemical Parameters and Liver Biomarkers.
Journal of clinical medicineHereditary Tyrosinemia Type 1 in Jordan: A Retrospective Study.
International journal of pediatricsFamily planning decisional needs assessment for recessive hereditary disorders: Insights from carrier couples and professionals.
Patient education and counselingFumarylacetoacetate hydrolase gene as a knockout target for hepatic chimerism and donor liver production.
Stem cell reportsEvaluation of dynamic thiol/disulfide homeostasis in hereditary tyrosinemia type 1 patients.
Pediatric researchCasein Glycomacropeptide: An Alternative Protein Substitute in Tyrosinemia Type I.
NutrientsNitisinone treatment during two pregnancies and breastfeeding in a woman with tyrosinemia type 1 - a case report.
Journal of pediatric endocrinology & metabolism : JPEMDiagnostic and Therapeutic Challenges of Hereditary Tyrosinemia Type 1 in Lebanon: A 12-Year Retrospective Review.
Frontiers in pediatricsLiver Transplantation: A Safe and Definitive Alternative to Lifelong Nitisinone for Tyrosinemia Type 1.
Indian journal of pediatricsInfluence of nitisinone and its metabolites on l-tyrosine metabolism in a model system.
ChemosphereSyndrome of Inappropriate Antidiuretic Hormone Secretion in a Patient with Uncontrolled Tyrosinaemia Type 1.
Sultan Qaboos University medical journalThe clinical variations and diagnostic challenges of deoxyguanosine kinase deficiency: a descriptive case series.
Journal of pediatric endocrinology & metabolism : JPEMTreatment adherence in tyrosinemia type 1 patients.
Orphanet journal of rare diseasesAdenine base editing and prime editing of chemically derived hepatic progenitors rescue genetic liver disease.
Cell stem cellTyrosinemia type 1 in pediatric nephrology: Not always straightforward.
Archives de pediatrie : organe officiel de la Societe francaise de pediatrieOutcome of Tyrosinemia Type 1 in Indian Children.
Journal of clinical and experimental hepatologyPhenotype, genotype, and outcome of 25 Palestinian patients with hereditary tyrosinemia type 1.
Metabolism openOxidative Stress, Glutathione Metabolism, and Liver Regeneration Pathways Are Activated in Hereditary Tyrosinemia Type 1 Mice upon Short-Term Nitisinone Discontinuation.
GenesHereditary Tyrosinemia Compounded With Hyperinsulinemic Hypoglycemia: Challenging Diagnosis of a Rare Case.
CureusCase report: Maternal tyrosinemia type 1a under NTBC treatment with tyrosine- and phenylalanine restricted diet in Chile.
American journal of medical genetics. Part C, Seminars in medical geneticsHepatocellular neoplasms arising in genetic metabolic disorders: steatosis is common in both the tumor and background liver.
Human pathologyThe future of gene-targeted therapy for hereditary tyrosinemia type 1 as a lead indication among the inborn errors of metabolism.
Expert opinion on orphan drugsBase editing: a brief review and a practical example.
Journal of biomedical researchEx Vivo Cell Therapy by Ectopic Hepatocyte Transplantation Treats the Porcine Tyrosinemia Model of Acute Liver Failure.
Molecular therapy. Methods & clinical developmentThe Importance of Succinylacetone: Tyrosinemia Type I Presenting with Hyperinsulinism and Multiorgan Failure Following Normal Newborn Screening.
International journal of neonatal screeningSevere neurological crisis in adult patients with Tyrosinemia type 1.
Annals of clinical and translational neurologyInduced Liver Regeneration Enhances CRISPR/Cas9-Mediated Gene Repair in Tyrosinemia Type 1.
Human gene therapyAspartame and Phe-Containing Degradation Products in Soft Drinks across Europe.
NutrientsLaboratory monitoring of patients with hereditary tyrosinemia type I.
Molecular genetics and metabolismAmelioration of an Inherited Metabolic Liver Disease through Creation of a De Novo Start Codon by Cytidine Base Editing.
Molecular therapy : the journal of the American Society of Gene TherapyInter-laboratory analytical improvement of succinylacetone and nitisinone quantification from dried blood spot samples.
JIMD reportsDried blood spot versus venous blood sampling for phenylalanine and tyrosine.
Orphanet journal of rare diseasesLate Development of Hepatocellular Carcinoma in Tyrosinemia Type 1 Despite Nitisinone (NTBC) Treatment.
Journal of pediatric gastroenterology and nutritionEmotional and behavioral problems, quality of life and metabolic control in NTBC-treated Tyrosinemia type 1 patients.
Orphanet journal of rare diseasesThe Effect of Various Doses of Phenylalanine Supplementation on Blood Phenylalanine and Tyrosine Concentrations in Tyrosinemia Type 1 Patients.
NutrientsLong-Term Outcomes and Practical Considerations in the Pharmacological Management of Tyrosinemia Type 1.
Paediatric drugsTyrosinemia Type 1 and symptoms of ADHD: Biochemical mechanisms and implications for treatment and prognosis.
American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric GeneticsBlood and Brain Biochemistry and Behaviour in NTBC and Dietary Treated Tyrosinemia Type 1 Mice.
NutrientsGenetic Analysis of Tyrosinemia Type 1 and Fructose-1, 6 Bisphosphatase Deficiency Affected in Pakistani Cohorts.
Fetal and pediatric pathologyA robust bacterial assay for high-throughput screening of human 4-hydroxyphenylpyruvate dioxygenase inhibitors.
Scientific reportsEvolution of tyrosinemia type 1 disease in patients treated with nitisinone in Spain.
MedicineHeme as an initial treatment for severe decompensation in tyrosinemia type 1.
Genetics in medicine : official journal of the American College of Medical GeneticsMutational spectrum of Mexican patients with tyrosinemia type 1: In silico modeling and predicted pathogenic effect of a novel missense FAH variant.
Molecular genetics & genomic medicineRevisiting hereditary tyrosinemia Type 1-spectrum of radiological findings.
BJR case reportsBiomarkers of Micronutrients in Regular Follow-Up for Tyrosinemia Type 1 and Phenylketonuria Patients.
NutrientsValidation of a therapeutic range for nitisinone in patients treated for tyrosinemia type 1 based on reduction of succinylacetone excretion.
JIMD reportsInterference of hydroxyphenylpyruvic acid, hydroxyphenyllactic acid and tyrosine on routine serum and urine clinical chemistry assays; implications for biochemical monitoring of patients with alkaptonuria treated with nitisinone.
Clinical biochemistryImaging liver nodules in tyrosinemia type-1: A retrospective review of 16 cases in a tertiary pediatric hospital.
European journal of radiologyEx Vivo Hepatocyte Reprograming Promotes Homology-Directed DNA Repair to Correct Metabolic Disease in Mice After Transplantation.
Hepatology communicationsPresence of three mutations in the fumarylacetoacetate hydrolase gene in a patient with atypical symptoms of hereditary tyrosinemia type I.
Molecular genetics and metabolismHepatotoxicity and Toxicology of In Vivo Lentiviral Vector Administration in Healthy and Liver-Injury Mouse Models.
Human gene therapy. Clinical developmentMedical care of patients with disorders of aromatic amino acid metabolism: a report based on the Polish National Health Fund data records.
Pediatric endocrinology, diabetes, and metabolismDiagnosis and the importance of early treatment of tyrosinemia type 1: A case report.
Clinical mass spectrometry (Del Mar, Calif.)Gene Editing Successfully Corrects 2 Amino Acid Disorders: In 2 preclinical studies using CRISPR-mediated gene editing, phenylketonuria and hereditary tyrosinemia type 1 were corrected.
American journal of medical genetics. Part AAutologous Gene and Cell Therapy Provides Safe and Long-Term Curative Therapy in A Large Pig Model of Hereditary Tyrosinemia Type 1.
Cell transplantationNon randomized study on the potential of nitisinone to inhibit cytochrome P450 2C9, 2D6, 2E1 and the organic anion transporters OAT1 and OAT3 in healthy volunteers.
European journal of clinical pharmacologyThe Unique Spectrum of Mutations in Patients with Hereditary Tyrosinemia Type 1 in Different Regions of the Russian Federation.
JIMD reportsDiscontinuation of NTBC after liver transplantation in tyrosinemia type 1.
Pediatrics international : official journal of the Japan Pediatric SocietyDendrimer-Based Lipid Nanoparticles Deliver Therapeutic FAH mRNA to Normalize Liver Function and Extend Survival in a Mouse Model of Hepatorenal Tyrosinemia Type I.
Advanced materials (Deerfield Beach, Fla.)In utero CRISPR-mediated therapeutic editing of metabolic genes.
Nature medicineCurative Ex Vivo Hepatocyte-Directed Gene Editing in a Mouse Model of Hereditary Tyrosinemia Type 1.
Human gene therapyNuclease-Mediated Gene Therapies for Inherited Metabolic Diseases of the Liver.
The Yale journal of biology and medicineNeurological Crises after Discontinuation of Nitisinone (NTBC) Treatment in Tyrosinemia.
Iranian journal of child neurologySimultaneous quantification of succinylacetone and nitisinone for therapeutic drug monitoring in the treatment of Tyrosinemia type 1.
Journal of chromatography. B, Analytical technologies in the biomedical and life sciencesDaily variation of NTBC and its relation to succinylacetone in tyrosinemia type 1 patients comparing a single dose to two doses a day.
Journal of inherited metabolic diseaseCaregiver Quality of Life with Tyrosinemia Type 1.
Journal of genetic counselingPresumptive brain influx of large neutral amino acids and the effect of phenylalanine supplementation in patients with Tyrosinemia type 1.
PloS oneEvaluation of pre-symptomatic nitisinone treatment on long-term outcomes in Tyrosinemia type 1 patients: a systematic review.
Orphanet journal of rare diseasesClinical utility of nitisinone for the treatment of hereditary tyrosinemia type-1 (HT-1).
The application of clinical geneticsHereditary Tyrosinemia Type 1 in Turkey.
Advances in experimental medicine and biologyLiver Cancer in Tyrosinemia Type 1.
Advances in experimental medicine and biologyNTBC and Correction of Renal Dysfunction.
Advances in experimental medicine and biologyThe Liver in Tyrosinemia Type I: Clinical Management and Course in Quebec.
Advances in experimental medicine and biologyMolecular Pathogenesis of Liver Injury in Hereditary Tyrosinemia 1.
Advances in experimental medicine and biologyMolecular Aspects of the FAH Mutations Involved in HT1 Disease.
Advances in experimental medicine and biologyBiochemical and Clinical Aspects of Hereditary Tyrosinemia Type 1.
Advances in experimental medicine and biologyComparison of a full systematic review versus rapid review approaches to assess a newborn screening test for tyrosinemia type 1.
Research synthesis methodsOpen-Label Single-Sequence Crossover Study Evaluating Pharmacokinetics, Efficacy, and Safety of Once-Daily Dosing of Nitisinone in Patients with Hereditary Tyrosinemia Type 1.
JIMD reports[Acute liver failure related to inherited metabolic diseases in young children].
Anales de pediatriaCase of hepatocellular carcinoma in a patient with hereditary tyrosinemia in the post-newborn screening era.
World journal of hepatologyLong-term cognitive functioning in individuals with tyrosinemia type 1 treated with nitisinone and protein-restricted diet.
Molecular genetics and metabolism reportsNewborn screening for Tyrosinemia type 1 using succinylacetone - a systematic review of test accuracy.
Orphanet journal of rare diseasesWhat Is the Best Blood Sampling Time for Metabolic Control of Phenylalanine and Tyrosine Concentrations in Tyrosinemia Type 1 Patients?
JIMD reportsLong-term outcome of expanded newborn screening at Boston children's hospital: benefits and challenges in defining true disease.
Journal of inherited metabolic diseaseFumarylacetoacetate Hydrolase Knock-out Rabbit Model for Hereditary Tyrosinemia Type 1.
The Journal of biological chemistryHypersuccinylacetonaemia and normal liver function in maleylacetoacetate isomerase deficiency.
Journal of medical geneticsChronic Phenotype Characterization of a Large-Animal Model of Hereditary Tyrosinemia Type 1.
The American journal of pathologyPediatric hepatocellular carcinoma in a developing country: Is the etiology changing?
Pediatric transplantationThe outcome of seven patients with hereditary tyrosinemia type 1.
Journal of pediatric endocrinology & metabolism : JPEMA novel homozygous mutation causing hereditary tyrosinemia type I in yakut patient in russia: case report.
Wiadomosci lekarskie (Warsaw, Poland : 1960)Curative ex vivo liver-directed gene therapy in a pig model of hereditary tyrosinemia type 1.
Science translational medicineNeurocognitive outcome in tyrosinemia type 1 patients compared to healthy controls.
Orphanet journal of rare diseasesIdentification of circulating microRNAs during the liver neoplastic process in a murine model of hereditary tyrosinemia type 1.
Scientific reportsTyrosinemia type 1 and irreversible neurologic crisis after one month discontinuation of nitisone.
Metabolic brain diseasePrenatal Diagnosis of Tyrosinemia Type 1 Using Next Generation Sequencing.
Fetal and pediatric pathologyAn unfortunate challenge: Ketogenic diet for the treatment of Lennox-Gastaut syndrome in tyrosinemia type 1.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyIron overload in hereditary tyrosinemia type 1 induces liver injury through the Sp1/Tfr2/hepcidin axis.
Journal of hepatology[Pharmacological and clinical profile of nitisinone (Orfadin(®) Capsules): a therapeutic agent for hereditary tyrosinemia type 1].
Nihon yakurigaku zasshi. Folia pharmacologica JaponicaDirect sequencing of FAH gene in Pakistani tyrosinemia type 1 families reveals a novel mutation.
Journal of pediatric endocrinology & metabolism : JPEMSustained NRF2 activation in hereditary leiomyomatosis and renal cell cancer (HLRCC) and in hereditary tyrosinemia type 1 (HT1).
Biochemical Society transactionsMolecular changes associated with chronic liver damage and neoplastic lesions in a murine model of hereditary tyrosinemia type 1.
Biochimica et biophysica actaThe Inhibitory Activity of Plants from Central Argentina on p-Hydroxyphenylpyruvate Dioxygenase. Isolation and Mechanism of Inhibition of a Flavanone from Flourensia oolepis.
Planta medicaClinical and Biochemical Profile of Tyrosinemia Type 1 in Tunisia.
Clinical laboratoryInherited metabolic disorders presenting as acute liver failure in newborns and young children: King's College Hospital experience.
European journal of pediatricsGeographical and Ethnic Distribution of Mutations of the Fumarylacetoacetate Hydrolase Gene in Hereditary Tyrosinemia Type 1.
JIMD reportsHepatocellular carcinoma in tyrosinemia type 1 without clear increase of AFP.
PediatricsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Tirosinemia tipo 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Tirosinemia tipo 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- SIADH as an Underrecognized Manifestation of Porphyria-like Crises in Hereditary Tyrosinemia Type 1: Clinical and Pathophysiological Insights.
- Identification and Characterization of Novel Variants of Fumarylacetoacetate Hydrolase (FAH) Gene in Clinically Suspected Patients of Tyrosinemia Type 1: Tertiary Care Centre Study of North India.
- Quantitative Succinylacetone Measurement by Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) Facilitates Diagnosis, Monitoring, and Characterization of Tyrosinemia Type 1 and Other Hypersuccinylacetonemias.
- Fusion Peptide-Incorporated Lipid Nanoparticles Boost Endosomal Escape and Enhance Cytosolic mRNA Delivery.
- Management of porphyria-like syndrome in tyrosinemia type 1.
- Two Years of Expanded Newborn Screening in Russia: High-Throughput Detection of Inherited Metabolic Disorders by Tandem Mass Spectrometry with Next-Generation Sequencing Confirmation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:882(Orphanet)
- OMIM OMIM:276700(OMIM)
- MONDO:0010161(MONDO)
- GARD:2658(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1747726(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Tirosinemia tipo 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA
- Reposicionamento
- fonte: Drug Repurposing Hub
- Ensaios clínicos
- fonte: ClinicalTrials.gov