A Síndrome de Sheldon-Hall (SHS) é uma condição rara, presente desde o nascimento, que causa encurtamento ou rigidez em diversas articulações. Ela é caracterizada por: encurtamento ou rigidez nas articulações mais distantes dos braços e pernas (como as das mãos e pés), rosto triangular, olhos com as aberturas voltadas para baixo, boca pequena e céu da boca alto e arqueado.
Introdução
O que você precisa saber de cara
A Síndrome de Sheldon-Hall (SHS) é uma condição rara, presente desde o nascimento, que causa encurtamento ou rigidez em diversas articulações. Ela é caracterizada por: encurtamento ou rigidez nas articulações mais distantes dos braços e pernas (como as das mãos e pés), rosto triangular, olhos com as aberturas voltadas para baixo, boca pequena e céu da boca alto e arqueado.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 19 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 48 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
5 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Not applicable.
Muscle contraction
Cytoplasm, myofibril
Arthrogryposis, distal, 2A
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA2A is characterized by contractures of the hands and feet, oropharyngeal abnormalities, scoliosis, and a distinctive face that includes a very small oral orifice, puckered lips, and a H-shaped dimple of the chin.
Voltage-gated ion channel responsible for the resting Na(+) permeability that controls neuronal excitability (PubMed:17448995, PubMed:31409833). NALCN channel functions as a multi-protein complex, which consists at least of NALCN, NALF1, UNC79 and UNC80 (PubMed:32494638, PubMed:33203861). NALCN is the voltage-sensing, pore-forming subunit of the NALCN channel complex (PubMed:17448995). NALCN channel complex is constitutively active and conducts monovalent cations but is blocked by physiological
Cell membrane
Hypotonia, infantile, with psychomotor retardation and characteristic facies 1
A neurodegenerative disease characterized by variable degrees of hypotonia, speech impairment, intellectual disability, pyramidal signs, subtle facial dysmorphism, and chronic constipation. Some patients manifest neuroaxonal dystrophy, optic atrophy, unmyelinated axons and spheroid bodies in tissue biopsies.
Binds to actin filaments in muscle and non-muscle cells. Plays a central role, in association with the troponin complex, in the calcium dependent regulation of vertebrate striated muscle contraction. Smooth muscle contraction is regulated by interaction with caldesmon. In non-muscle cells is implicated in stabilizing cytoskeleton actin filaments. The non-muscle isoform may have a role in agonist-mediated receptor internalization
Cytoplasm, cytoskeleton
Congenital myopathy 23
An autosomal dominant muscular disorder characterized clinically by hypotonia and muscle weakness, and a static or slowly progressive clinical course. Disease onset ranges from birth to childhood. Histologic examination of muscle fibers shows various anomalies including fiber type disproportion, an irregular myofibrillar network, abnormal thread-like or rod-shaped structures, and cap-like structures which are well demarcated and peripherally located under the sarcolemma with abnormal accumulation of sarcomeric proteins.
Troponin I is the inhibitory subunit of troponin, the thin filament regulatory complex which confers calcium-sensitivity to striated muscle actomyosin ATPase activity
Arthrogryposis, distal, 2B1
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. DA2B is characterized by contractures of the hands and feet, and a distinctive face characterized by prominent nasolabial folds, small mouth and downslanting palpebral fissures. DA2B1 inheritance is autosomal dominant.
Troponin T is the tropomyosin-binding subunit of troponin, the thin filament regulatory complex which confers calcium-sensitivity to striated muscle actomyosin ATPase activity
Arthrogryposis, distal, 2B2
A form of distal arthrogryposis, a disease characterized by congenital joint contractures that mainly involve two or more distal parts of the limbs, in the absence of a primary neurological or muscle disease. Distal arthrogryposis type 2 is characterized by contractures of the hands and feet, and a distinctive face characterized by prominent nasolabial folds, small mouth and downslanting palpebral fissures. DA2B2 inheritance is autosomal dominant.
Variantes genéticas (ClinVar)
929 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 2 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Sheldon-Hall
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
2 ensaios clínicos encontrados.
Publicações mais relevantes
Bi-allelic variants in MYH3 cause recessively-inherited arthrogryposis.
Arthrogryposis is a clinical feature defined by congenital joint contractures in two or more different body areas which occurs in between 1/3000 and 1/5000 live births. Variants in multiple genes have been associated with distal arthrogryposis syndromes. Heterozygous variants in MYH3 have been identified to cause the dominantly-inherited distal arthrogryposis conditions, Freeman-Sheldon syndrome, Sheldon-Hall syndrome, and multiple pterygium syndrome. In contrast, MYH3 variants underlie both dominantly and recessively inherited Contractures, Pterygia, and Spondylocarpotarsal Fusion syndromes (CPSFS) which are characterized by extensive bony abnormalities in addition to congenital contractures. Here we report two affected sibs with distal arthrogryposis born to unaffected, distantly related parents. Sequencing revealed that both sibs were homozygous for two ultra-rare MYH3 variants, c.3445G>A (p.Glu1149Lys) and c.4760T>C (p.Leu1587Pro). Sequencing and deletion/duplication analysis of 169 other arthrogryposis genes yielded no other compelling candidate variants. This is the first report of biallelic variants in MYH3 being implicated in a distal arthrogryposis phenotype without the additional features of CPSFS. Thus, akin to CPSFS, both dominant and recessively inherited distal arthrogryposis can be caused by variants in MYH3.
Identification of two novel MYH3 variants causing different phenotypes in prenatal diagnosis.
The MYH3 gene encodes the embryonic myosin heavy chain, which is crucial for the skeletal and muscular development. The MYH3 variants are associated with distal arthrogryposis type 2A (Freeman-Sheldon syndrome), distal arthrogryposis type 2B3 (Sheldon-Hall syndrome), CPSFS1A (Contractures, pterygia, and spondylocarpostarsal fusion syndrome 1A) and CPSFS1B, which have some shared characteristics and great variability of clinical phenotypes. In this study, we report two novel MYH3 missense variants c.1024T>G (p.Phe342Val) and c.3872A>C (p.Gln1291Pro), demonstrating different phenotypes in the prenatal setting. This study expands the spectrum of MYH3 variants and supports the domain-specific genotype-phenotype correlation of MYH3.
Diagnostic work-up and phenotypic characteristics of a family with variable severity of distal arthrogryposis type 2B (Sheldon-Hall syndrome) and TNNT3 pathogenic variant.
Background: Sheldon-Hall syndrome (SHS) or distal arthrogryposis 2B (DA2B) is a rare clinically and genetically heterogeneous multiple congenital contracture syndrome characterized by contractures of the distal joints of the limbs and mild facial involvement, due to pathogenic variants in genes encoding the fast-twitch skeletal muscle contractile myofiber complex (TNNT3, TNNI2, TMP2, and MYH3 genes). Patients and methods: A 16-year-old boy with a history of congenital distal arthrogryposis developed severe kyphoscoliosis and respiratory insufficiency. His mother and younger sister had phenotypes compatible with SHS but to a much lesser extent. Diagnostic work-up included physical examination and whole-body muscular MRI (WBMRI) in all three patients and electroneuromyography (ENMG) and paravertebral muscle biopsy in the proband. DNA sequencing was used to confirm the diagnosis. Results: Physical examination suggested the diagnosis of SHS. No muscle signal abnormalities were found in WBMRI. Large motor unit potentials and reduced recruitment suggestive of neurogenic changes were observed on needle EMG in distal and paravertebral muscles in the proband. DNA sequencing revealed a pathogenic variant in TNNT3 (c.187C>T), which segregated as a dominant trait with the phenotype. Discussion: This is the first report on neurogenic features in a patient with DA2B and a pathogenic variant in TNNT3 encoding the fast-twitch skeletal muscle contractile myofiber complex. A superimposed length-dependent motor nerve involvement was unexpected. Whether developmental disarrangements in number, distribution, or innervation of the motor unit in fetal life might lead to pseudo-neurogenic EMG features warrants further studies, as well as the role of genetic modifiers in SHS variability.
Freeman-Sheldon Syndrome with Stiff Knee Gait - A Case Report.
Freeman-Sheldon syndrome (FSS), also known as the distal arthrogryposis (DA) type 2A, is a rare congenital anomaly. We report a unique case of the DA type 2A with mixed clinical features and the unusual presentation of bilateral congenital dislocation of the knee but had unassisted stiff knee gait. A 5-year-old female child presented to the clinic with the complaint of inability to bend both knees since birth. She had an unassisted bipedal gait, but could not squat, cross-leg sit, run, and climb stairs without assistance. Her youngest brother had a similar presentation but succumbed to death at the age of 5 months due to respiratory distress. Clinical features were in the favor of FSS. Her serum creatinine kinase level was normal and the electromyography of bilateral tibialis anterior and abductor pollicis brevis was not suggestive of the myotonia. Radiograph of the skull showed cooper beaten skull appearance whereas bilateral pelvis with the hip showed following changes in the right hip; decrease femoral epiphysis height, horizontal proximal femoral physis, and the coxa brevia. She was initially managed conservatively by weekly stretching, manipulation, and casting. As a result, she could flex her knee up to 20°. Although the quadricepsplasty might be helpful for the persistent extension deformity, there was marked quadriceps weakness which could make it harder for the child to stand and walk. In addition, the abnormal muscle physiology in FSS may result in unfavorable outcomes after the surgery. Moreover, a consideration of the surgical aspect is not free of risks which include difficult endotracheal intubation, vein access, and malignant hyperthermia. FSS is a rare congenital anomaly that should be differentiated from another syndrome of the close resemblance, Sheldon Hall syndrome and Schwartz Jampel syndrome which are other rare autosomal recessive disorders characterized by myotonia and the chondrodysplasia. Conservative management has still a role in bilateral knee involvement especially if the patient is an independent walker.
[Genetic analysis and prenatal diagnosis of a pregnant woman with Sheldon-Hall syndrome].
To provide genetic testing and prenatal diagnosis for a woman with Sheldon-Hall syndrome. The woman was subjected to targeted capture and next-generation sequencing for variant of genes associated with skeletal disorders. And the result was verified in her parents and fetus. The woman was found to harbor a c.188G>A variant of the TNNT3 gene, which was also found in her affected mother and the fetus. Her grandmother and grandmother's brother had similar manifestations, which was in line with an autosomal dominant inheritance. The same variant was not found in her father. The c.188G>A variant of the TNNT3 gene probably underlay the distal joint contracture in this pedigree, based on which prenatal diagnosis was attained.
Publicações recentes
Bi-allelic variants in MYH3 cause recessively-inherited arthrogryposis.
Identification of two novel MYH3 variants causing different phenotypes in prenatal diagnosis.
Diagnostic work-up and phenotypic characteristics of a family with variable severity of distal arthrogryposis type 2B (Sheldon-Hall syndrome) and TNNT3 pathogenic variant.
Freeman-Sheldon Syndrome with Stiff Knee Gait - A Case Report.
[Genetic analysis and prenatal diagnosis of a pregnant woman with Sheldon-Hall syndrome].
📚 EuropePMC10 artigos no totalmostrando 13
Bi-allelic variants in MYH3 cause recessively-inherited arthrogryposis.
Clinical geneticsIdentification of two novel MYH3 variants causing different phenotypes in prenatal diagnosis.
Prenatal diagnosisDiagnostic work-up and phenotypic characteristics of a family with variable severity of distal arthrogryposis type 2B (Sheldon-Hall syndrome) and TNNT3 pathogenic variant.
Frontiers in geneticsFreeman-Sheldon Syndrome with Stiff Knee Gait - A Case Report.
Journal of orthopaedic case reports[Genetic analysis and prenatal diagnosis of a pregnant woman with Sheldon-Hall syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsDrosophila myosin mutants model the disparate severity of type 1 and type 2B distal arthrogryposis and indicate an enhanced actin affinity mechanism.
Skeletal muscleFindings, Phenotypes, Diagnostic Accuracy, and Treatment in Freeman-Burian Syndrome.
The Journal of craniofacial surgeryMyosin heavy chain mutations that cause Freeman-Sheldon syndrome lead to muscle structural and functional defects in Drosophila.
Developmental biologyA MYH3 mutation identified for the first time in a Chinese family with Sheldon-Hall syndrome (DA2B).
Neuromuscular disorders : NMDA novel pathogenic MYH3 mutation in a child with Sheldon-Hall syndrome and vertebral fusions.
American journal of medical genetics. Part AFindings, phenotypes, and outcomes in Freeman-Sheldon and Sheldon-Hall syndromes and distal arthrogryposis types 1 and 3: protocol for systematic review and patient-level data meta-analysis.
Systematic reviewsFreeman-Sheldon syndrome in a 29-year-old woman presenting with rare and previously undescribed features.
BMJ case reportsThe clubfoot painted by Jusepe de Ribera: a controversial diagnosis.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Sheldon-Hall.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Sheldon-Hall
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Bi-allelic variants in MYH3 cause recessively-inherited arthrogryposis.
- Identification of two novel MYH3 variants causing different phenotypes in prenatal diagnosis.
- Diagnostic work-up and phenotypic characteristics of a family with variable severity of distal arthrogryposis type 2B (Sheldon-Hall syndrome) and TNNT3 pathogenic variant.
- Freeman-Sheldon Syndrome with Stiff Knee Gait - A Case Report.
- [Genetic analysis and prenatal diagnosis of a pregnant woman with Sheldon-Hall syndrome].Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics· 2020· PMID 32820522mais citado
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1147(Orphanet)
- MONDO:0011128(MONDO)
- GARD:16556(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q9390344(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
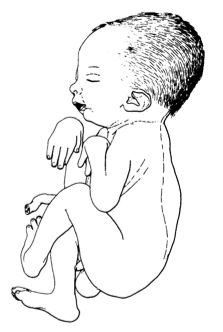
Síndrome Sheldon-Hall
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata