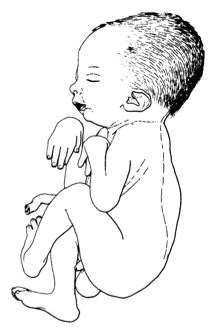
A síndrome de Barber Say (BSS) é uma displasia ectodérmica rara de início neonatal caracterizada por hipertricose congênita generalizada, pele atrófica, ectrópio e microstomia.
Introdução
O que você precisa saber de cara
A síndrome de Barber Say (BSS) é uma displasia ectodérmica rara de início neonatal caracterizada por hipertricose congênita generalizada, pele atrófica, ectrópio e microstomia.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 24 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 62 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Not applicable.
Curadoria gene-doença
fontes oficiaisBinds to the E-box consensus sequence 5'-CANNTG-3' as a heterodimer and inhibits transcriptional activation by MYOD1, MYOG, MEF2A and MEF2C. Also represses expression of pro-inflammatory cytokines such as TNFA and IL1B. Involved in postnatal glycogen storage and energy metabolism (By similarity). Inhibits the premature or ectopic differentiation of preosteoblast cells during osteogenesis, possibly by changing the internal signal transduction response of osteoblasts to external growth factors
NucleusCytoplasm
Focal facial dermal dysplasia 3, Setleis type
A form of focal facial dermal dysplasia, a group of developmental defects characterized by bitemporal or preauricular skin lesions resembling aplasia cutis congenita. FFDD3 is characterized by distinctive bitemporal scar-like depressions resembling forceps marks, and additional facial features, including a coarse and leonine appearance, absent eyelashes on both lids or multiple rows on the upper lids, absent Meibomian glands, slanted eyebrows, chin clefting, and hypo- or hyperpigmentation of the skin. Histologically, the bitemporal lesion is an ectodermal dysplasia with near absence of subcutaneous fat, suggesting insufficient migration of neural crest cells into the frontonasal process and the first branchial arch.
Variantes genéticas (ClinVar)
94 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Barber-Say
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Multiple rhabdomyomatous mesenchymal hamartomas in a patient with mosaic Barber-Say syndrome.
Barber-Say syndrome (BSS) is a rare congenital ectodermal dysplasia with few cases reported in the literature. We describe a 9-year-old boy with congenital generalized hypertrichosis and multiple rhabdomyomatous mesenchymal hamartomas (RMHs) on his nose and periocular region. Next-generation sequencing, performed in DNA from a blood sample, and RMH tissue, revealed a pathogenic variant in the TWIST2 gene, which was not detected in a salivary sample of the patient, nor in his parents. Therefore, we consider this variant as de novo mosaicism. To our knowledge, this is the first case of multiple RMHs associated with BSS.
High-power laser as a treatment of recurrent gingival fibromatosis in a patient with a rare syndrome: A case report.
Ocular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
Ablepharon-macrostomia syndrome (AMS) and Barber-Say syndrome (BSS) are congenital ectodermal dysplasias associated with mutations in the TWIST2 gene. Among the ophthalmic anomalies that occur in these syndromes, underdevelopment of the anterior lamella of the eyelid is a defining feature. Reports of mosaic expression of TWIST2 mutations are extremely rare, with only five confirmed or suspected cases described to date. Mosaic expression of TWIST2 variants is correlated with a less severe phenotype than that reported for the typical expression of TWIST2 variants associated with BSS or AMS. Abnormal development of the anterior lamella appears to be a common feature in all cases of AMS with mosaic expression. Here, we describe the phenotype of a patient with mosaic expression of a TWIST2 mutation that is typically associated with AMS. We additionally describe the surgical approach employed in the treatment of this patient.
Barber Say Syndrome (A New Case Report).
Barber Say syndrome (BSS) is a rare ectodermal dysplasia with neonatal onset characterized by congenital generalized hypertrichosis, atrophic skin, ectropion and macrostomia. A literature review showed less than 20 previously reported cases of Barber Say syndrome. This presentation reports a one day old female with syndrome face, low hairline, coarse face, macrostomia, thin upper lip, bilateral ectropion and hypertelorism, hypertrichosis, senile skin appearance, hypoplastic nipples and one area of mild skin atrophy. These findings are consistent with BSS.
Multidisciplinary eyelid reconstruction in Barber-Say syndrome: A case report.
Barber-Say syndrome is an unusual dysplasia caused by the mutation of the TWIST2 gene (2q37.3), which encodes a protein that acts at an epigenetic level. The case is presented of a 2-day-old male child in whom ectropion, hypertelorism, hypertrichosis and other dysmorphic features led to the clinical diagnosis of Barber-Say syndrome, which was later confirmed with genetic tests. Around 20 cases have been reported on this syndrome, of which less than half have described the surgical technique, as it represents a surgical challenge. The approach in this case included a lateral tarsorrhaphy and skin grafts taken from the volar surface of the forearm, retroauricular area and supraclavicular fossa, as well as autologous lipografts from the inner side of both thighs for palpebral reconstruction. This is the first case of Barber-Say syndrome in which the use of skin grafts are taken from supraclavicular fossa and forearms.
Publicações recentes
Multiple rhabdomyomatous mesenchymal hamartomas in a patient with mosaic Barber-Say syndrome.
High-power laser as a treatment of recurrent gingival fibromatosis in a patient with a rare syndrome: A case report.
Ocular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
Barber Say Syndrome (A New Case Report).
Multidisciplinary eyelid reconstruction in Barber-Say syndrome: A case report.
📚 EuropePMC19 artigos no totalmostrando 15
Multiple rhabdomyomatous mesenchymal hamartomas in a patient with mosaic Barber-Say syndrome.
Pediatric dermatologyHigh-power laser as a treatment of recurrent gingival fibromatosis in a patient with a rare syndrome: A case report.
Photodermatology, photoimmunology & photomedicineOcular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
Orbit (Amsterdam, Netherlands)Barber Say Syndrome (A New Case Report).
Indian dermatology online journalMultidisciplinary eyelid reconstruction in Barber-Say syndrome: A case report.
Archivos de la Sociedad Espanola de OftalmologiaAblepharon and craniosynostosis in a patient with a localized TWIST1 basic domain substitution.
American journal of medical genetics. Part AClinical Description, Molecular Analysis of TWIST2 Gene, and Surgical Treatment in a Patient With Barber-Say Syndrome.
Ophthalmic plastic and reconstructive surgeryBarber-Say Syndrome and Ablepharon-Macrostomia Syndrome: A Patient's View.
Molecular syndromologyBarber-say syndrome: a confirmed case of TWIST2 gene mutation.
Clinical case reportsThe focal facial dermal dysplasias: phenotypic spectrum and molecular genetic heterogeneity.
Journal of medical geneticsBarber-Say syndrome and Ablepharon-Macrostomia syndrome: An overview.
American journal of medical genetics. Part ATransmission of Barber-Say syndrome from a mosaic father to his child in an Indian family.
Clinical dysmorphologyGeneral anesthesia of a Japanese infant with Barber-Say syndrome: a case report.
JA clinical reportsRecurrent Mutations in the Basic Domain of TWIST2 Cause Ablepharon Macrostomia and Barber-Say Syndromes.
American journal of human geneticsA case of Barber-Say syndrome in a male Japanese newborn.
Clinical case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Barber-Say.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Barber-Say
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Multiple rhabdomyomatous mesenchymal hamartomas in a patient with mosaic Barber-Say syndrome.
- High-power laser as a treatment of recurrent gingival fibromatosis in a patient with a rare syndrome: A case report.
- Ocular adnexal phenotype and management of a patient with mosaic expression of a mutation in TWIST2.
- Barber Say Syndrome (A New Case Report).
- Multidisciplinary eyelid reconstruction in Barber-Say syndrome: A case report.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1231(Orphanet)
- OMIM OMIM:209885(OMIM)
- MONDO:0008853(MONDO)
- GARD:819(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q18616565(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Barber-Say
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata