A síndrome de Curry-Jones é uma forma de craniossinostose sindrômica, caracterizada por craniossinostose coronal unilateral ou sinostose de sutura múltipla associada à agenesia completa ou parcial do corpo caloso, polissindactilia pré-axial e sindactilia das mãos e/ou pés, juntamente com anomalias da pele (áreas brancas peroladas características que se tornam cicatrizes e atróficas, crescimento anormal de pêlos ao redor dos olhos e/ou bochechas, e em membros), olhos (colobomas de íris, microftalmia) e intestino (intestino curto congênito, má rotação, dismotilidade, constipação crônica, sangramento e miofibromas). Atraso no desenvolvimento e graus variáveis de deficiência intelectual também podem ser observados. Foram relatados múltiplos hamartomas intra-abdominais do músculo liso, tricoblastoma da pele, meningoceles occipitais e desenvolvimento de meduloblastoma desmoplásico.
Introdução
O que você precisa saber de cara
A síndrome de Curry-Jones é uma forma de craniossinostose sindrômica, caracterizada por craniossinostose coronal unilateral ou sinostose de sutura múltipla associada à agenesia completa ou parcial do corpo caloso, polissindactilia pré-axial e sindactilia das mãos e/ou pés, juntamente com anomalias da pele (áreas brancas peroladas características que se tornam cicatrizes e atróficas, crescimento anormal de pêlos ao redor dos olhos e/ou bochechas, e em membros), olhos (colobomas de íris, microftalmia) e intestino (intestino curto congênito, má rotação, dismotilidade, constipação crônica, sangramento e miofibromas). Atraso no desenvolvimento e graus variáveis de deficiência intelectual também podem ser observados. Foram relatados múltiplos hamartomas intra-abdominais do músculo liso, tricoblastoma da pele, meningoceles occipitais e desenvolvimento de meduloblastoma desmoplásico.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 12 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 45 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Not applicable.
Curadoria gene-doença
fontes oficiaisFlavoenzyme which catalyzes the oxidation of spermine to spermidine. Can also use N(1)-acetylspermine and spermidine as substrates, with different affinity depending on the isoform (isozyme) and on the experimental conditions. Plays an important role in the regulation of polyamine intracellular concentration and has the potential to act as a determinant of cellular sensitivity to the antitumor polyamine analogs. May contribute to beta-alanine production via aldehyde dehydrogenase conversion of 3
CytoplasmNucleus
Variantes genéticas (ClinVar)
59 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 8 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
2 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Curry-Jones
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Occurrence and characterization of medulloblastoma in a patient with Curry-Jones syndrome.
Medulloblastoma in a Patient with Curry-Jones Syndrome with a mosaic variant, c.1234C > T (p.Leu412Phe), in SMO.
Happle-Tinschert, Curry-Jones and segmental basal cell naevus syndromes, overlapping disorders caused by somatic mutations in hedgehog signalling genes: the mosaic hedgehog spectrum.
Happle-Tinschert syndrome (HTS) and Curry-Jones syndrome (CJS; OMIM 601707) are rare, sporadic, multisystem disorders characterized by hypo- and hyperpigmented skin patches following Blaschko's lines, plus acral skeletal and other abnormalities. The blaschkoid pattern implies mosaicism, and indeed CJS was found in 2016 to be caused by a recurrent postzygotic mutation in a gene of the hedgehog signalling pathway, namely SMO, c.1234C>T, p.Leu412Phe. More recently the original case of HTS was found to carry the same somatic mutation. Despite this genetic and phenotypic overlap, two significant differences remained between the two syndromes. The histological hallmark of HTS, basaloid follicular hamartomas, is not a feature of CJS. Meanwhile, the severe gastrointestinal manifestations regularly reported in CJS had not been described in HTS. We report a patient whose phenotype was entirely consistent with HTS apart from intractable constipation, and a second patient with classic features of CJS plus early-onset medulloblastoma, a feature of basal cell naevus syndrome (BCNS). Both had the same recurrent SMO mutation. This prompted a literature review that revealed a case with the same somatic mutation, with basaloid follicular hamartomas and other features of both CJS and BCNS. Segmental BCNS can also be caused by a somatic mutation in PTCH1. We thus demonstrate for the first time phenotypic and genetic overlap between HTS, CJS and segmental BCNS. All of these conditions are caused by somatic mutations in genes of the hedgehog signalling pathway and we therefore propose the unifying term 'mosaic hedgehog spectrum'. What's already known about this topic? Happle-Tinschert syndrome (HTS) and Curry-Jones syndrome (CJS) are rare mosaic multisystem disorders with linear skin lesions. CJS is characterized by severe constipation, which has not previously been reported in HTS. HTS is characterized by basaloid follicular hamartomas, which are not a recognized feature of CJS. The recurrent mosaic SMO mutation found in CJS was recently reported in a patient with HTS. What does this study add? We describe a patient with HTS and intractable constipation, and a case of CJS with medulloblastoma. Both patients had the same recurrent somatic SMO mutation also found in a case reported as segmental basal cell naevus syndrome. SMO functions in the hedgehog pathway, explaining phenotypic overlap between HTS, CJS and mosaic basal cell naevus syndrome. We propose the term 'mosaic hedgehog spectrum' for these overlapping conditions.
Gastrointestinal disorders in Curry-Jones syndrome: Clinical and molecular insights from an affected newborn.
Curry-Jones syndrome (CJS) is a pattern of malformation that includes craniosynostosis, pre-axial polysyndactyly, agenesis of the corpus callosum, cutaneous and gastrointestinal abnormalities. A recurrent, mosaic mutation of SMO (c.1234 C>T; p.Leu412Phe) causes CJS. This report describes the gastrointestinal and surgical findings in a baby with CJS who presented with abdominal obstruction and reviews the spectrum of gastrointestinal malformations in this rare disorder. A 41-week, 4,165 g, female presented with craniosynostosis, pre-axial polysyndactyly, and cutaneous findings consistent with a clinical diagnosis of CJS. The infant developed abdominal distension beginning on the second day of life. Surgical exploration revealed an intestinal malrotation for which she underwent a Ladd procedure. Multiple small nodules were found on the surface of the small and large bowel in addition to an apparent intestinal duplication that seemed to originate posterior to the pancreas. Histopathology of serosal nodules revealed bundles of smooth muscle with associated ganglion cells. Molecular analysis demonstrated the SMO c.1234 C>T mutation in varying amounts in affected skin (up to 35%) and intestinal hamartoma (26%). Gastrointestinal features including structural malformations, motility disorders, and upper GI bleeding are major causes of morbidity in CJS. Smooth muscle hamartomas are a recognized feature of children with CJS typically presenting with abdominal obstruction requiring surgical intervention. A somatic mutation in SMO likely accounts for the structural malformations and predisposition to form bowel hamartomas and myofibromas. The mutation burden in the involved tissues likely accounts for the variable manifestations.
A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome.
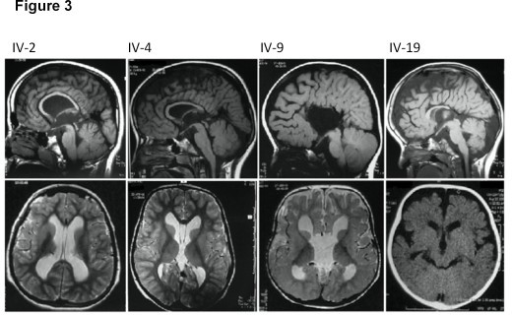
Curry-Jones syndrome (CJS) is a multisystem disorder characterized by patchy skin lesions, polysyndactyly, diverse cerebral malformations, unicoronal craniosynostosis, iris colobomas, microphthalmia, and intestinal malrotation with myofibromas or hamartomas. Cerebellar medulloblastoma has been described in a single affected individual; in another, biopsy of skin lesions showed features of trichoblastoma. The combination of asymmetric clinical features, patchy skin manifestations, and neoplastic association previously led to the suggestion that this could be a mosaic condition, possibly involving hedgehog (Hh) signaling. Here, we show that CJS is caused by recurrent somatic mosaicism for a nonsynonymous variant in SMO (c.1234C>T [p.Leu412Phe]), encoding smoothened (SMO), a G-protein-coupled receptor that transduces Hh signaling. We identified eight mutation-positive individuals (two of whom had not been reported previously) with highly similar phenotypes and demonstrated varying amounts of the mutant allele in different tissues. We present detailed findings from brain MRI in three mutation-positive individuals. Somatic SMO mutations that result in constitutive activation have been described in several tumors, including medulloblastoma, ameloblastoma, and basal cell carcinoma. Strikingly, the most common of these mutations is the identical nonsynonymous variant encoding p.Leu412Phe. Furthermore, this substitution has been shown to activate SMO in the absence of Hh signaling, providing an explanation for tumor development in CJS. This raises therapeutic possibilities for using recently generated Hh-pathway inhibitors. In summary, our work uncovers the major genetic cause of CJS and illustrates strategies for gene discovery in the context of low-level tissue-specific somatic mosaicism.
Happle-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome: reply from the authors.
Publicações recentes
Occurrence and characterization of medulloblastoma in a patient with Curry-Jones syndrome.
🥈 ObservacionalHapple-Tinschert, Curry-Jones and segmental basal cell naevus syndromes, overlapping disorders caused by somatic mutations in hedgehog signalling genes: the mosaic hedgehog spectrum.
📖 RevisãoGastrointestinal disorders in Curry-Jones syndrome: Clinical and molecular insights from an affected newborn.
Happle-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome: reply from the authors.
Happle-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome.
📚 EuropePMC8 artigos no totalmostrando 6
Occurrence and characterization of medulloblastoma in a patient with Curry-Jones syndrome.
Clinical geneticsHapple-Tinschert, Curry-Jones and segmental basal cell naevus syndromes, overlapping disorders caused by somatic mutations in hedgehog signalling genes: the mosaic hedgehog spectrum.
The British journal of dermatologyGastrointestinal disorders in Curry-Jones syndrome: Clinical and molecular insights from an affected newborn.
American journal of medical genetics. Part AHapple-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome: reply from the authors.
The British journal of dermatologyHapple-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome.
The British journal of dermatologyA Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome.
American journal of human geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Curry-Jones.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Curry-Jones
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Occurrence and characterization of medulloblastoma in a patient with Curry-Jones syndrome.
- Happle-Tinschert, Curry-Jones and segmental basal cell naevus syndromes, overlapping disorders caused by somatic mutations in hedgehog signalling genes: the mosaic hedgehog spectrum.
- Gastrointestinal disorders in Curry-Jones syndrome: Clinical and molecular insights from an affected newborn.
- A Recurrent Mosaic Mutation in SMO, Encoding the Hedgehog Signal Transducer Smoothened, Is the Major Cause of Curry-Jones Syndrome.
- Happle-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome: reply from the authors.
- Happle-Tinschert syndrome can be caused by a mosaic SMO mutation and is suggested to be a variant of Curry-Jones syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1553(Orphanet)
- OMIM OMIM:601707(OMIM)
- MONDO:0011134(MONDO)
- GARD:5584(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783221(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Curry-Jones
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata