A displasia septoóptica (SOD) é um distúrbio clinicamente heterogêneo caracterizado pela tríade clássica de hipoplasia do nervo óptico, anormalidades hormonais hipofisárias e defeitos cerebrais na linha média.
Introdução
O que você precisa saber de cara
A displasia septoóptica (SOD) é um distúrbio clinicamente heterogêneo caracterizado pela tríade clássica de hipoplasia do nervo óptico, anormalidades hormonais hipofisárias e defeitos cerebrais na linha média.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 39 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
7 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive, Multigenic/multifactorial, Not applicable.
Curadoria gene-doença
fontes oficiaisReceptor for prokineticin 2. Exclusively coupled to the G(q) subclass of heteromeric G proteins. Activation leads to mobilization of calcium, stimulation of phosphoinositide turnover and activation of p44/p42 mitogen-activated protein kinase
Cell membrane
Hypogonadotropic hypogonadism 3 with or without anosmia
A disorder characterized by absent or incomplete sexual maturation by the age of 18 years, in conjunction with low levels of circulating gonadotropins and testosterone and no other abnormalities of the hypothalamic-pituitary axis. In some cases, it is associated with non-reproductive phenotypes, such as anosmia, cleft palate, and sensorineural hearing loss. Anosmia or hyposmia is related to the absence or hypoplasia of the olfactory bulbs and tracts. Hypogonadism is due to deficiency in gonadotropin-releasing hormone and probably results from a failure of embryonic migration of gonadotropin-releasing hormone-synthesizing neurons. In the presence of anosmia, idiopathic hypogonadotropic hypogonadism is referred to as Kallmann syndrome, whereas in the presence of a normal sense of smell, it has been termed normosmic idiopathic hypogonadotropic hypogonadism (nIHH).
Tyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of embryonic development, cell proliferation, differentiation and migration. Required for normal mesoderm patterning and correct axial organization during embryonic development, normal skeletogenesis and normal development of the gonadotropin-releasing hormone (GnRH) neuronal system. Phosphorylates PLCG1, FRS2, GAB1 and SHB. Ligand binding leads to the activati
Cell membraneNucleusCytoplasm, cytosolCytoplasmic vesicle
Pfeiffer syndrome
A syndrome characterized by the association of craniosynostosis, broad and deviated thumbs and big toes, and partial syndactyly of the fingers and toes. Three subtypes are known: mild autosomal dominant form (type 1); cloverleaf skull, elbow ankylosis, early death, sporadic (type 2); craniosynostosis, early demise, sporadic (type 3).
Required for the normal development of the forebrain, eyes and other anterior structures such as the olfactory placodes and pituitary gland. Possible transcriptional repressor. Binds to the palindromic PIII sequence, 5'-AGCTTGAGTCTAATTGAATTAACTGTAC-3'. HESX1 and PROP1 bind as heterodimers on this palindromic site, and, in vitro, HESX1 can antagonize PROP1 activation
Nucleus
Septooptic dysplasia
A clinically heterogeneous disorder defined by any combination of optic nerve hypoplasia, pituitary gland hypoplasia with panhypopopituitarism, and midline abnormalities of the brain, including absence of the corpus callosum and septum pellucidum.
Transcription factor required during the formation of the hypothalamo-pituitary axis. May function as a switch in neuronal development. Keeps neural cells undifferentiated by counteracting the activity of proneural proteins and suppresses neuronal differentiation. Required also within the pharyngeal epithelia for craniofacial morphogenesis. Controls a genetic switch in male development. Is necessary for initiating male sex determination by directing the development of supporting cell precursors
Nucleus
Panhypopituitarism X-linked
Affected individuals have absent infundibulum, anterior pituitary hypoplasia, and ectopic posterior pituitary.
Transcription factor that forms a trimeric complex with OCT4 on DNA and controls the expression of a number of genes involved in embryonic development such as YES1, FGF4, UTF1 and ZFP206 (By similarity). Binds to the proximal enhancer region of NANOG (By similarity). Critical for early embryogenesis and for embryonic stem cell pluripotency (PubMed:18035408). Downstream SRRT target that mediates the promotion of neural stem cell self-renewal (By similarity). Keeps neural cells undifferentiated by
Nucleus speckleCytoplasmNucleus
Microphthalmia, syndromic, 3
A disease characterized by the rare association of malformations including uni- or bilateral anophthalmia or microphthalmia, and esophageal atresia with trachoesophageal fistula. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Transcription factor that plays a role in the development of the hypothalamo-pituitary axis, postnatal brain growth, and visual and renal function (PubMed:24022475). Specifically recognizes the xenobiotic response element (XRE)
Nucleus
Webb-Dattani syndrome
A disorder characterized by postnatal microcephaly with fronto-temporal lobe hypoplasia, multiple pituitary hormone deficiency, global developmental delay, seizures, severe visual impairment and abnormalities of the kidneys and urinary tract.
Transcription factor probably involved in the development of the brain and the sense organs. Can bind to the bicoid/BCD target sequence (BTS): 5'-TCTAATCCC-3'
Nucleus
Microphthalmia, syndromic, 5
Patients manifest unilateral or bilateral microphthalmia/clinical anophthalmia and variable additional features including pituitary dysfunction, coloboma, microcornea, cataract, retinal dystrophy, hypoplasia or agenesis of the optic nerve, agenesis of the corpus callosum, developmental delay, joint laxity, hypotonia, and seizures. Microphthalmia is a disorder of eye formation, ranging from small size of a single eye to complete bilateral absence of ocular tissues (anophthalmia). In many cases, microphthalmia/anophthalmia occurs in association with syndromes that include non-ocular abnormalities.
Variantes genéticas (ClinVar)
335 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
42 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Espectro clínico de displasia septo-óptica
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
5 ensaios clínicos encontrados, 1 ativos.
Publicações mais relevantes
Partial ectopic posterior pituitary: A rare imaging entity with literature review.
Abnormal development of the posterior pituitary gland can lead to an ectopic location of the neurohypophysis, commonly seen at the median eminence of the hypothalamus or along the infundibular stalk. A partial ectopic posterior pituitary (PEPP) is a very rare variant of the ectopic posterior pituitary, defined as the presence of a double bright spot of neurohypophysis seen in both orthotopic and ectopic locations. We report a two-year-old male toddler with bilateral optic nerve hypoplasia and severe visual impairment who presented to the endocrine outpatient clinic for hypopituitarism evaluation. Magnetic resonance imaging (MRI) of the brain revealed a hypoplastic pituitary gland and infundibulum with a double bright spot of neurohypophysis in the expected normal location and along the median eminence. Severe hypoplasia of both optic nerves and the optic chiasm was also seen. Septum pellucidum was present with no evidence of other brain malformations. The findings are in the septo-optic dysplasia spectrum associated with hypothalamic-pituitary dysfunction and a very rare entity called PEPP. To our knowledge, only a handful of reported cases of this rare entity exist in the literature.
Neurodevelopmental impairments in children with septo-optic dysplasia spectrum conditions: a systematic review.
Septo-optic dysplasia (SOD) is a rare condition diagnosed in children with two or more of the following: hypopituitarism, midline brain abnormalities, and optic nerve hypoplasia. Children with SOD experience varied visual impairment and endocrine dysfunction. Autistic-like behaviours have been reported; however, their nature and prevalence remain to be fully understood. The present systematic review aimed to explore the type and prevalence of neurodevelopmental impairments in children with SOD spectrum conditions. The search was conducted in PubMed, EMBASE, and PsycInfo. Hand-searching reference lists of included studies was conducted. All peer-reviewed, observational studies assessing behavioural and cognitive impairments or autism spectrum disorder (ASD) symptoms in children (< 18 years) with SOD, optic nerve hypoplasia, and SOD-plus were included. Studies were excluded if they did not report standardised measures of neurodevelopmental impairments or ASD outcomes. From 2132 screened articles, 20 articles reporting data from a total of 479 children were included in prevalence estimates. Of 14 studies assessing cognitive-developmental outcomes, 175 of 336 (52%) children presented with intellectual disability or developmental delay. A diagnosis of ASD or clinical level of symptoms was observed in 65 of 187 (35%) children across five studies. Only five studies assessed for dysfunction across behavioural, emotional, or social domains and reported impairments in 88 of 184 (48%) of children assessed. Importantly, high heterogeneity among the samples in relation to their neuroanatomical, endocrine, and optic nerve involvement meant that it was not possible to statistically assess the relative contribution of these confounding factors to the specific neurodevelopmental phenotype. This was further limited by the variation in study designs and behavioural assessments used across the included studies, which may have increased the risk of information bias. This systematic review suggests that the prevalence of neurodevelopmental impairments in children within the SOD spectrum may be high. Clinicians should therefore consider including formal assessments of ASD symptoms and neurodevelopmental impairments alongside routine care. There is, additionally, a need for further research to define and validate a standardised battery of tools that accurately identify neurodevelopmental impairments in SOD spectrum conditions, and for research to identify the likely causal mechanisms.
The phenotypic spectrum associated with OTX2 mutations in humans.
The transcription factor OTX2 is implicated in ocular, craniofacial, and pituitary development. We aimed to establish the contribution of OTX2 mutations in congenital hypopituitarism patients with/without eye abnormalities, study functional consequences, and establish OTX2 expression in the human brain, with a view to investigate the mechanism of action. We screened patients from the UK (n = 103), international centres (n = 24), and Brazil (n = 282); 145 were within the septo-optic dysplasia spectrum, and 264 had no eye phenotype. Transactivation ability of OTX2 variants was analysed in murine hypothalamic GT1-7 neurons. In situ hybridization was performed on human embryonic brain sections. Genetically engineered mice were generated with a series of C-terminal OTX2 variants. Two chromosomal deletions and six haploinsufficient mutations were identified in individuals with eye abnormalities; an affected relative of one patient harboured the same mutation without an ocular phenotype. OTX2 truncations led to significant transactivation reduction. A missense variant was identified in another patient without eye abnormalities; however, studies revealed it was most likely not causative. In the mouse, truncations proximal to aa219 caused anophthalmia, while distal truncations and the missense variant were tolerated. During human embryogenesis, OTX2 was expressed in the posterior pituitary, retina, ear, thalamus, choroid plexus, and partially in the hypothalamus, but not in the anterior pituitary. OTX2 mutations are rarely associated with hypopituitarism in isolation without eye abnormalities, and may be variably penetrant, even within the same pedigree. Our data suggest that the endocrine phenotypes in patients with OTX2 mutations are of hypothalamic origin.
Increasing incidence of optic nerve hypoplasia/septo-optic dysplasia spectrum: Geographic clustering in Northern Canada.
Owing to the shared embryonic origin, defects in development of optic nerves are often seen in conjunction with defects affecting the surrounding brain and pituitary gland. Optic nerve hypoplasia (ONH) and septo-optic dysplasia (SOD) represent a clinical spectrum associated with visual, pituitary and severe central nervous system structural abnormalities (SODplus). Based on changing clinical patterns, our primary objective was to examine trends in annual incidence of ONH/SOD and geographical clustering in Manitoba. This was a retrospective 1996 to 2015 chart review with extraction of anthropometric measures, radiologic findings, parental characteristics, endocrinopathies and neurologic symptoms from all involved in care. Postal codes were used to assign map co-ordinates and identify relevant census-based deprivation indices. Ninety-three children were identified in our catchment area; Poisson regression confirmed a striking 1.11-fold annual increase (95% confidence interval 1.07 to 1.16) or ~800% over two decades. The annual incidence (averaged 2010 to 2014 chart data) reached 53.3 per 100,000, affecting 1 in 1875 live births. Most (~55%) had SODplus. Common presenting features were hypoglycemia, nystagmus, seizures and developmental delay; 40% had hormone deficiencies; 80% had reduced visual acuity, typically bilateral. Many were premature with young, primiparous mothers. Unhealthy maternal lifestyles and severe material deprivation were noted. There was disproportionate clustering in individuals from Northern Manitoba at three times the average provincial rate. We noted a dramatic rise in the annual incidence of ONH/SOD, which was strongly associated with poverty and northern communities. The pattern was consistent with environmental or nutritional etiologies. Many children were severely affected with increased morbidity and health care burdens.
Publicações recentes
Partial ectopic posterior pituitary: A rare imaging entity with literature review.
Neurodevelopmental impairments in children with septo-optic dysplasia spectrum conditions: a systematic review.
The phenotypic spectrum associated with OTX2 mutations in humans.
Increasing incidence of optic nerve hypoplasia/septo-optic dysplasia spectrum: Geographic clustering in Northern Canada.
Midbrain-hindbrain involvement in septo-optic dysplasia.
📚 EuropePMC3 artigos no totalmostrando 4
Partial ectopic posterior pituitary: A rare imaging entity with literature review.
The neuroradiology journalNeurodevelopmental impairments in children with septo-optic dysplasia spectrum conditions: a systematic review.
Molecular autismThe phenotypic spectrum associated with OTX2 mutations in humans.
European journal of endocrinologyIncreasing incidence of optic nerve hypoplasia/septo-optic dysplasia spectrum: Geographic clustering in Northern Canada.
Paediatrics & child healthAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Espectro clínico de displasia septo-óptica.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Espectro clínico de displasia septo-óptica
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Partial ectopic posterior pituitary: A rare imaging entity with literature review.
- Neurodevelopmental impairments in children with septo-optic dysplasia spectrum conditions: a systematic review.
- The phenotypic spectrum associated with OTX2 mutations in humans.
- Increasing incidence of optic nerve hypoplasia/septo-optic dysplasia spectrum: Geographic clustering in Northern Canada.
- Midbrain-hindbrain involvement in septo-optic dysplasia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3157(Orphanet)
- OMIM OMIM:182230(OMIM)
- MONDO:0008428(MONDO)
- GARD:7627(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q2756703(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
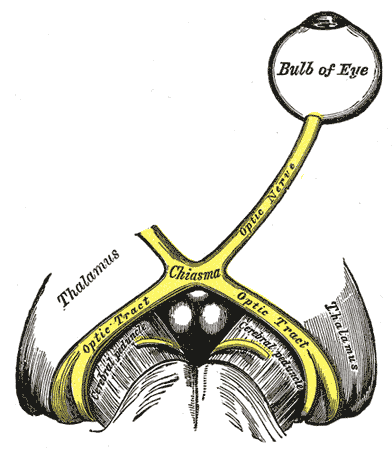
Espectro clínico de displasia septo-óptica
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov