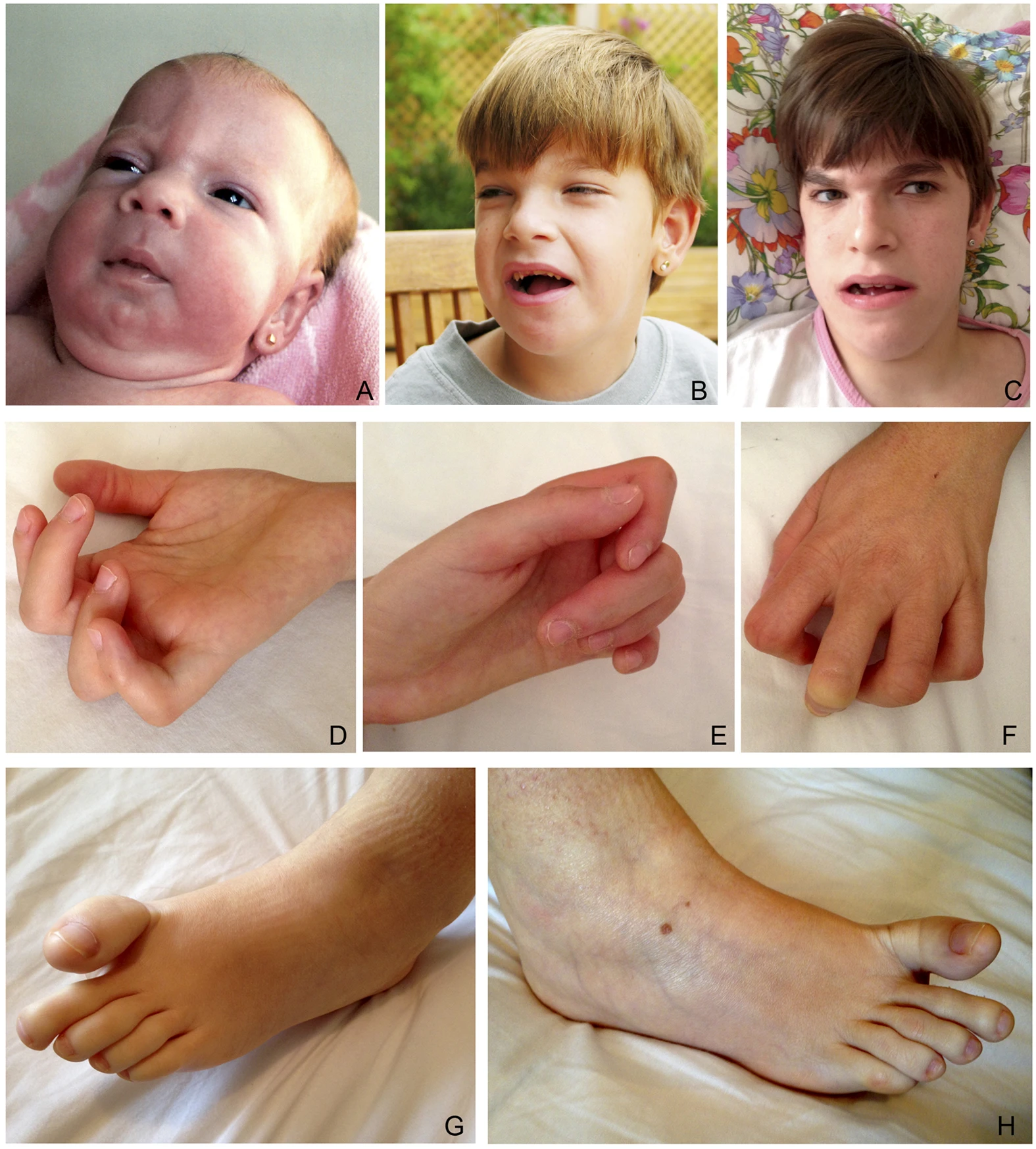
A síndrome de Fountain é uma doença genética muito rara que afeta várias partes do corpo. Ela é caracterizada por deficiência intelectual, surdez, alterações nos ossos e traços faciais mais acentuados.
Introdução
O que você precisa saber de cara
A síndrome de Fountain é uma doença genética muito rara que afeta várias partes do corpo. Ela é caracterizada por deficiência intelectual, surdez, alterações nos ossos e traços faciais mais acentuados.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 17 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 49 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Nenhum gene associado encontrado
Os dados genéticos desta condição ainda estão sendo catalogados.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Fountain
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Integrative epigenetic and transcriptomic profiling of whole blood and fibroblasts in Hao-Fountain syndrome.
Hao-Fountain syndrome (HAFOUS) is a rare autosomal dominant neurodevelopmental disorder caused by pathogenic USP7 variants. A diagnostic blood DNA methylation episignature has been established, yet the broader regulatory consequences of USP7 haploinsufficiency and their tissue specificity remain incompletely characterized. We performed genome-wide DNA methylation profiling, RNA sequencing, and cis expression quantitative trait methylation (eQTM) analysis in whole blood (n = 9) and patient-derived skin fibroblasts (n = 4). Differential methylation was assessed and methylation-expression coupling within ±250 kb of each DMR. DMRs were further interpreted using BCOR, H2AK119ub1, and H3K27me3 ChIP-Rx datasets from neural models. Blood reproduced the established USP7 hypermethylation episignature and yielded 17 significant DMRs, accompanied by modest numbers of differentially expressed genes and eQTMs. Fibroblasts displayed internally coherent regulatory patterns, including 2,143 nominal DMRs, 310 differentially expressed genes, and 559 significant eQTMs. Convergent methylation-expression changes prominently involved the HOXB cluster (HOXB3, HOXB5, HOXB6). Both blood- and fibroblast-derived DMRs showed significant enrichment for BCOR- and H2AK119ub1-marked regions, consistent with disruption of non-canonical PRC1.1-associated chromatin. Cross-tissue comparison revealed limited overlap, supporting marked tissue specificity in methylation-expression relationships. USP7 haploinsufficiency is associated with a restricted set of regulatory loci enriched within PRC1-associated chromatin domains. Fibroblasts revealed coherent methylation and expression changes at developmental genes, whereas blood captured the diagnostic episignature and a smaller set of downstream regulatory alterations. Together, this dual-tissue integrative analysis refines the molecular consequences of reduced USP7 dosage and provides a framework for future mechanistic studies in disease-relevant cellular models.
Generation of two human induced pluripotent stem cell lines (iPSC) from patients with Hao-Fountain Syndrome.
Hao-Fountain Syndrome (HAFOUS) is an autosomal dominant neurodevelopmental disorder caused by pathogenic variants in the USP7 gene. Research into the molecular mechanisms of HAFOUS has been limited by the lack of suitable human model systems. In this study, we introduce induced pluripotent stem cell (iPSC) lines derived from a HAFOUS patient, providing a valuable model to investigate mechanisms of altered cell lineage commitment during disease progression. This model offers a platform to explore the role of USP7 variants in HAFOUS pathogenesis and develop potential therapeutic strategies.
Small-molecule allosteric activator of ubiquitin-specific protease 7 (USP7).
Ubiquitin-specific protease 7 (USP7) is a deubiquitylase essential for cell homeostasis, DNA repair, and regulation of both tumor suppressors and oncogenes. Recently, haploinsufficiency of USP7 has been associated with Hao-Fountain syndrome (HAFOUS), a rare neurodevelopmental disorder. Although a range of USP7 inhibitors have been developed over the last decade, in the context of HAFOUS, USP7 activators may represent a more relevant approach. To address this challenge, we report the identification and characterization of a small-molecule activator of USP7 called MS-8. Structural and functional studies show that MS-8 allosterically activates USP7, mimicking the endogenous autoactivation mechanism of the enzyme. We observed that MS-8 engages and activates mutant USP7 in a cellular context, impacting downstream proteins. Taken together, our study provides validation of the USP7 activator that paves the way toward activation-driven USP7 pharmacology.
Functional spectrum of USP7 pathogenic variants in Hao-Fountain syndrome: Insights into the enzyme's activity, stability, and allosteric modulation.
Hao-Fountain syndrome is a rare neurodevelopmental disorder caused by mutations in the deubiquitinating enzyme Ubiquitin-Specific Protease 7 (USP7). Due to the novelty of the disease and its poorly understood molecular mechanisms, treatments for the syndrome are currently lacking. This study examines the effects of 11 patient-derived variants located within the catalytic domain of USP7, focusing on their impact on the enzyme's activity, thermodynamic stability, and substrate recognition. Our findings reveal a spectrum of functional consequences, ranging from complete inactivation to hyperactivation of USP7. Notably, we identify a specific subset of pathogenic variants whose catalytic activity can be significantly boosted using an allosteric activator, MS-8. These results provide insight into USP7 malfunction in Hao-Fountain syndrome-linked variants and pave the way for improved prognostic approaches and targeted treatments in the future.
Activation dynamics of ubiquitin-specific protease 7.
Ubiquitin-specific protease 7 (USP7) is a deubiquitinating enzyme that plays a crucial role in cellular processes, including the maintenance of genome stability and regulation of antiviral and immune responses. Its dysfunction is linked to various cancers and neurodevelopmental disorders such as Hao-Fountain syndrome. Unlike other USP-family enzymes, the triad of catalytic residues in USP7 adopts an inactive conformation and undergoes rearrangement into the active state upon substrate binding. Despite its potential importance for regulating the enzyme's activity, the dynamics of USP7 have not been explored. In this study, we combine advanced CPMG NMR relaxation dispersion measurements with the analysis of enzyme kinetics to investigate the conformational dynamics of USP7 in solution and its role in enzyme activation. Our results suggest that apo-USP7 exists in a dynamic equilibrium, transiently switching between inactive and low-populated active conformations, indicating that enzyme activation can occur spontaneously, even in the absence of a substrate. Furthermore, we show that the Hao-Fountain syndrome-associated variant G392D enhances the conformational dynamics of the enzyme, leading to a significant increase in its catalytic activity. This study captures the sparsely populated, "invisible" active conformation of USP7 and demonstrates how changes in enzyme dynamics can contribute to activity, offering broader insights into enzyme function and disease mechanisms.
Publicações recentes
Integrative epigenetic and transcriptomic profiling of whole blood and fibroblasts in Hao-Fountain syndrome.
Generation of two human induced pluripotent stem cell lines (iPSC) from patients with Hao-Fountain Syndrome.
Nanopore sequencing enables combined detection of USP7 variants and a known Hao-Fountain syndrome episignature.
USP7-Related Hao-Fountain Syndrome.
Small-molecule allosteric activator of ubiquitin-specific protease 7 (USP7).
📚 EuropePMC20 artigos no totalmostrando 18
Integrative epigenetic and transcriptomic profiling of whole blood and fibroblasts in Hao-Fountain syndrome.
Frontiers in cell and developmental biologyGeneration of two human induced pluripotent stem cell lines (iPSC) from patients with Hao-Fountain Syndrome.
Stem cell researchNanopore sequencing enables combined detection of USP7 variants and a known Hao-Fountain syndrome episignature.
Frontiers in geneticsThe spectrum of communication abilities in children with 12 rare neurodevelopmental disorders: a qualitative study with caregivers.
Journal of child psychology and psychiatry, and allied disciplinesSmall-molecule allosteric activator of ubiquitin-specific protease 7 (USP7).
Proceedings of the National Academy of Sciences of the United States of AmericaFunctional spectrum of USP7 pathogenic variants in Hao-Fountain syndrome: Insights into the enzyme's activity, stability, and allosteric modulation.
Proceedings of the National Academy of Sciences of the United States of AmericaNeurodevelopmental disorder: Hao-Fountain syndrome with USP7 mutation-a case report.
Journal of medical case reportsActivation dynamics of ubiquitin-specific protease 7.
Proceedings of the National Academy of Sciences of the United States of AmericaSertraline and Astemizole Enhance the Deubiquitinase Activity of USP7 by Binding to Its Switching Loop Region.
Journal of medicinal chemistryHao-Fountain syndrome protein USP7 controls neuronal differentiation via BCOR-ncPRC1.1.
Genes & developmentThe Hao-Fountain syndrome protein USP7 regulates neuronal connectivity in the brain via a novel p53-independent ubiquitin signaling pathway.
Cell reportsIdentification of Two Variants c.2697A > C and c.3305A > C in USP7 by Analysis of Whole-Exome Sequencing in Chinese Patients with Hao-Fountain Syndrome.
Global medical geneticsHao-Fountain syndrome: 32 novel patients reveal new insights into the clinical spectrum.
Clinical geneticsDNA methylation episignature, extension of the clinical features, and comparative epigenomic profiling of Hao-Fountain syndrome caused by variants in USP7.
Genetics in medicine : official journal of the American College of Medical GeneticsA Rare Case of Hao-Fountain Syndrome Mimicking Fragile X Syndrome.
CureusExpansion of the mutation spectrum and phenotype of USP7-related neurodevelopmental disorder.
Frontiers in molecular neuroscienceHao-Fountain syndrome and genital disorders: report of a new possible association.
Italian journal of pediatricsComplex Presentation of Hao-Fountain Syndrome Solved by Exome Sequencing Highlighting Co-Occurring Genomic Variants.
GenesAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Fountain.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Fountain
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Integrative epigenetic and transcriptomic profiling of whole blood and fibroblasts in Hao-Fountain syndrome.
- Generation of two human induced pluripotent stem cell lines (iPSC) from patients with Hao-Fountain Syndrome.
- Small-molecule allosteric activator of ubiquitin-specific protease 7 (USP7).Proceedings of the National Academy of Sciences of the United States of America· 2025· PMID 41086218mais citado
- Functional spectrum of USP7 pathogenic variants in Hao-Fountain syndrome: Insights into the enzyme's activity, stability, and allosteric modulation.Proceedings of the National Academy of Sciences of the United States of America· 2025· PMID 40982686mais citado
- Activation dynamics of ubiquitin-specific protease 7.Proceedings of the National Academy of Sciences of the United States of America· 2025· PMID 40397674mais citado
- Nanopore sequencing enables combined detection of USP7 variants and a known Hao-Fountain syndrome episignature.
- USP7-Related Hao-Fountain Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:3219(Orphanet)
- OMIM OMIM:229120(OMIM)
- MONDO:0009241(MONDO)
- GARD:64(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q5474830(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Fountain
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata