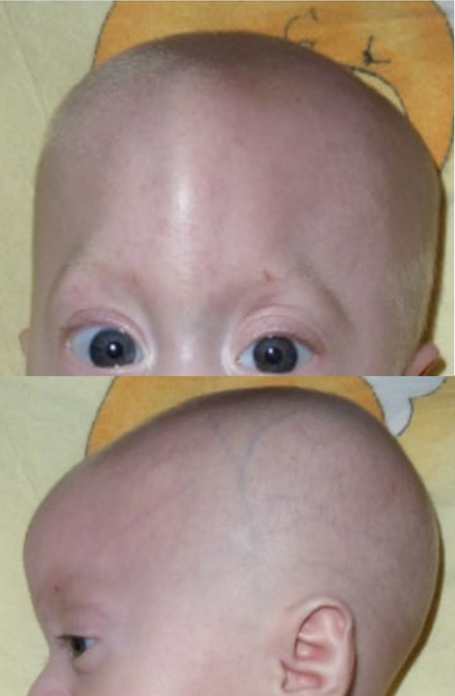
A síndrome oral-facio-digital, tipo 9, é caracterizada por palato altamente arqueado com língua bífida e caninos inferiores supranumerários bilaterais, língua hamartomatosa, frênulas múltiplas, hipertelorismo, telecanto, estrabismo, ponta nasal larga e/ou bífida, baixa estatura, hálux bífidos, metatarso bifurcado, poli e sindactilia, déficit intelectual leve e anormalidades específicas da retina (coloboma bilateral do disco óptico e retinal). displasia com descolamento parcial).
Introdução
O que você precisa saber de cara
A síndrome oral-facio-digital, tipo 9, é caracterizada por palato altamente arqueado com língua bífida e caninos inferiores supranumerários bilaterais, língua hamartomatosa, frênulas múltiplas, hipertelorismo, telecanto, estrabismo, ponta nasal larga e/ou bífida, baixa estatura, hálux bífidos, metatarso bifurcado, poli e sindactilia, déficit intelectual leve e anormalidades específicas da retina (coloboma bilateral do disco óptico e retinal). displasia com descolamento parcial).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 8 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 25 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisRequired for high-level Shh responses in the developing neural tube. Together with CDK20, controls the structure of the primary cilium by coordinating assembly of the ciliary membrane and axoneme, allowing GLI2 to be properly activated in response to Shh signaling (By similarity)
CytoplasmCell projection, cilium
Orofaciodigital syndrome 9
A form of orofaciodigital syndrome, a group of heterogeneous disorders characterized by malformations of the oral cavity, face and digits, and associated phenotypic abnormalities that lead to the delineation of various subtypes. OFD9 is an autosomal recessive form characterized by a variable phenotype. Clinical features are midline defects, including abnormal pituitary development that results in variable pituitary hormone deficiencies, facial dysmorphic features including frontal bossing and hypertelorism, and variable eye defects including microphthalmia, coloboma, and retinal dystrophy. Affected individuals manifest variable psychomotor development.
Variantes genéticas (ClinVar)
65 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 1.638 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome orofaciodigital tipo 9
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mostrando amostra de 5 publicações de um total de 50
Publicações recentes
RNA Sequencing for Rare Disease Diagnosis in a South African Family: A Novel Exon Elongation Event in OFD1.
The luminal ring protein C2CD3 acts as a radial in-to-out organizer of the distal centriole and appendages.
Expanding the phenotype associated with biallelic SCNM1 variants.
Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
The Luminal Ring Protein C2CD3 Acts as a Radial In-to-Out Organizer of the Distal Centriole and Appendages.
📚 EuropePMC86 artigos no totalmostrando 0
Ver todos os 86 no EuropePMCAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome orofaciodigital tipo 9.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome orofaciodigital tipo 9
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- RNA Sequencing for Rare Disease Diagnosis in a South African Family: A Novel Exon Elongation Event in OFD1.
- The luminal ring protein C2CD3 acts as a radial in-to-out organizer of the distal centriole and appendages.
- Expanding the phenotype associated with biallelic SCNM1 variants.
- Missense Variants in the Second Transmembrane Domain of TMEM17 Disrupt Its Stability and Function and Lead to a Wide Phenotypic Spectrum of Ciliopathies.
- The Luminal Ring Protein C2CD3 Acts as a Radial In-to-Out Organizer of the Distal Centriole and Appendages.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:141007(Orphanet)
- OMIM OMIM:258865(OMIM)
- MONDO:0009795(MONDO)
- GARD:10520(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q21154050(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome orofaciodigital tipo 9
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata