Distúrbio genético que afeta o metabolismo de ácidos graxos e a produção de energia, resultando em sintomas neurológicos, cardíacos e musculares. A deficiência na produção ou transporte de carnitina prejudica a gliconeogênese e pode levar a rabdomiólise e insuficiência renal.
Introdução
O que você precisa saber de cara
Visão geral
A alteração do ciclo da carnitina e do transporte de carnitina é um grupo de doenças genéticas raras que afetam a forma como o corpo utiliza a carnitina para transformar gordura em energia. A carnitina é uma substância essencial para levar as gorduras para dentro das mitocôndrias (as “usinas de energia” das células), onde são queimadas para gerar energia. Quando esse processo não funciona corretamente, o corpo pode ter dificuldade em produzir energia, especialmente durante períodos de jejum ou doença. Isso pode levar a uma série de problemas de saúde, como fraqueza muscular, problemas cardíacos e alterações no metabolismo.[1][3]
Sinais e sintomas
Os sinais e sintomas podem variar muito de pessoa para pessoa, mesmo dentro da mesma família. Alguns dos sintomas mais comuns incluem: convulsões, vômitos, dores musculares (mialgia), cãibras musculares que pioram com exercício, fraqueza, batimento cardíaco lento (bradicardia), insuficiência cardíaca congestiva, parada cardiorrespiratória, acúmulo de gordura no fígado (esteatose hepática macrovesicular), níveis elevados de enzimas do fígado (alanina aminotransferase e aspartato aminotransferase), problemas renais (doença renal crônica estágio 5, insuficiência renal, oligúria), alterações nos gânglios da base do cérebro (incluindo cistos), hidrocefalia, nistagmo (movimentos oculares involuntários), palato ogival (céu da boca alto e estreito), congestão nasal, hiperlipidemia transitória (aumento temporário de gorduras no sangue) e gliconeogênese prejudicada (dificuldade do corpo em produzir glicose).[1][3]
Causas genéticas
Essas condições são causadas por alterações (mutações) em genes que fornecem instruções para proteínas envolvidas no ciclo da carnitina e no transporte de carnitina. Os genes conhecidos por estarem associados a essas doenças são: CPT1A (carnitina O-palmitoiltransferase 1, isoforma hepática), CPT2 (carnitina O-palmitoiltransferase 2, mitocondrial), SLC25A20 (proteína transportadora mitocondrial de carnitina/acilcarnitina) e SLC22A5 (transportador 2 de cátions orgânicos/carnitina). Mutações nesses genes podem prejudicar a capacidade do corpo de transportar ou utilizar a carnitina, levando aos sintomas descritos.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica, exames laboratoriais (como dosagem de carnitina no sangue e urina, e perfil de acilcarnitinas) e confirmação por testes genéticos. Atualmente, existem 336 testes genéticos disponíveis para essas condições, e mais de 1.492 variantes genéticas diferentes já foram registradas no banco de dados ClinVar. O diagnóstico genético pode ajudar a identificar a causa exata e orientar o aconselhamento familiar.[1][4]
Tratamento e manejo
O tratamento é individualizado e pode incluir suplementação de carnitina (L-carnitina), dieta com baixo teor de gordura e rica em carboidratos, e evitar jejuns prolongados. O manejo também envolve monitoramento regular da função cardíaca, hepática e renal, além de acompanhamento com equipe multidisciplinar (médicos geneticistas, nutricionistas, cardiologistas, nefrologistas). Não há cobertura específica pelo Sistema Único de Saúde (SUS) para essas condições no momento.[1]
Tratamentos citados na literatura
Não há tratamentos específicos citados na literatura científica disponíveis nos dados fornecidos.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade da mutação genética e a precocidade do diagnóstico e tratamento. Com manejo adequado, muitas pessoas conseguem ter uma qualidade de vida razoável, mas complicações como insuficiência cardíaca, renal ou hepática podem reduzir a expectativa de vida. O acompanhamento médico regular é essencial para prevenir crises metabólicas e tratar complicações precocemente.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Distúrbio genético que afeta o metabolismo de ácidos graxos e a produção de energia, resultando em sintomas neurológicos, cardíacos e musculares. A deficiência na produção ou transporte de carnitina prejudica a gliconeogênese e pode levar a rabdomiólise e insuficiência renal.
Tem tratamento?
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A alteração do ciclo da carnitina e do transporte de carnitina é um grupo de doenças genéticas raras que afetam a forma como o corpo utiliza a carnitina para transformar gordura em energia. A carnitina é uma substância essencial para levar as gorduras para dentro das mitocôndrias (as “usinas de energia” das células), onde são queimadas para gerar energia. Quando esse processo não funciona corretamente, o corpo pode ter dificuldade em produzir energia, especialmente durante períodos de jejum ou doença. Isso pode levar a uma série de problemas de saúde, como fraqueza muscular, problemas cardíacos e alterações no metabolismo.[1][3]
Sinais e sintomas
Os sinais e sintomas podem variar muito de pessoa para pessoa, mesmo dentro da mesma família. Alguns dos sintomas mais comuns incluem: convulsões, vômitos, dores musculares (mialgia), cãibras musculares que pioram com exercício, fraqueza, batimento cardíaco lento (bradicardia), insuficiência cardíaca congestiva, parada cardiorrespiratória, acúmulo de gordura no fígado (esteatose hepática macrovesicular), níveis elevados de enzimas do fígado (alanina aminotransferase e aspartato aminotransferase), problemas renais (doença renal crônica estágio 5, insuficiência renal, oligúria), alterações nos gânglios da base do cérebro (incluindo cistos), hidrocefalia, nistagmo (movimentos oculares involuntários), palato ogival (céu da boca alto e estreito), congestão nasal, hiperlipidemia transitória (aumento temporário de gorduras no sangue) e gliconeogênese prejudicada (dificuldade do corpo em produzir glicose).[1][3]
Causas genéticas
Essas condições são causadas por alterações (mutações) em genes que fornecem instruções para proteínas envolvidas no ciclo da carnitina e no transporte de carnitina. Os genes conhecidos por estarem associados a essas doenças são: CPT1A (carnitina O-palmitoiltransferase 1, isoforma hepática), CPT2 (carnitina O-palmitoiltransferase 2, mitocondrial), SLC25A20 (proteína transportadora mitocondrial de carnitina/acilcarnitina) e SLC22A5 (transportador 2 de cátions orgânicos/carnitina). Mutações nesses genes podem prejudicar a capacidade do corpo de transportar ou utilizar a carnitina, levando aos sintomas descritos.[1][4]
Diagnóstico
O diagnóstico é baseado na avaliação clínica, exames laboratoriais (como dosagem de carnitina no sangue e urina, e perfil de acilcarnitinas) e confirmação por testes genéticos. Atualmente, existem 336 testes genéticos disponíveis para essas condições, e mais de 1.492 variantes genéticas diferentes já foram registradas no banco de dados ClinVar. O diagnóstico genético pode ajudar a identificar a causa exata e orientar o aconselhamento familiar.[1][4]
Tratamento e manejo
O tratamento é individualizado e pode incluir suplementação de carnitina (L-carnitina), dieta com baixo teor de gordura e rica em carboidratos, e evitar jejuns prolongados. O manejo também envolve monitoramento regular da função cardíaca, hepática e renal, além de acompanhamento com equipe multidisciplinar (médicos geneticistas, nutricionistas, cardiologistas, nefrologistas). Não há cobertura específica pelo Sistema Único de Saúde (SUS) para essas condições no momento.[1]
Tratamentos citados na literatura
Não há tratamentos específicos citados na literatura científica disponíveis nos dados fornecidos.[1]
Prognóstico e qualidade de vida
O prognóstico varia conforme a gravidade da mutação genética e a precocidade do diagnóstico e tratamento. Com manejo adequado, muitas pessoas conseguem ter uma qualidade de vida razoável, mas complicações como insuficiência cardíaca, renal ou hepática podem reduzir a expectativa de vida. O acompanhamento médico regular é essencial para prevenir crises metabólicas e tratar complicações precocemente.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 56 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 171 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição.
Catalyzes the transfer of the acyl group of long-chain fatty acid-CoA conjugates onto carnitine, an essential step for the mitochondrial uptake of long-chain fatty acids and their subsequent beta-oxidation in the mitochondrion (PubMed:11350182, PubMed:14517221, PubMed:16651524, PubMed:9691089). Also possesses a lysine succinyltransferase activity that can regulate enzymatic activity of substrate proteins such as ENO1 and metabolism independent of its classical carnitine O-palmitoyltransferase ac
Mitochondrion outer membrane
Carnitine palmitoyltransferase 1A deficiency
Rare autosomal recessive metabolic disorder of long-chain fatty acid oxidation characterized by severe episodes of hypoketotic hypoglycemia usually occurring after fasting or illness. Onset is in infancy or early childhood.
Involved in the intramitochondrial synthesis of acylcarnitines from accumulated acyl-CoA metabolites (PubMed:20538056, PubMed:24780397). Reconverts acylcarnitines back into the respective acyl-CoA esters that can then undergo beta-oxidation, an essential step for the mitochondrial uptake of long-chain fatty acids and their subsequent beta-oxidation in the mitochondrion. Active with medium (C8-C12) and long-chain (C14-C18) acyl-CoA esters (PubMed:20538056)
Mitochondrion inner membrane
Carnitine palmitoyltransferase 2 deficiency, myopathic, stress-induced
An autosomal recessive disorder of mitochondrial long-chain fatty acid oxidation, characterized by recurrent myoglobinuria, episodes of muscle pain, stiffness, and rhabdomyolysis. These symptoms are exacerbated by prolonged exercise, fasting, cold, or viral infection. CPT2DM affects most frequently children or young adults, and severity of attacks is highly variable. Myoglobinuria can cause kidney failure and death.
Mediates the electroneutral exchange of acylcarnitines (O-acyl-(R)-carnitine or L-acylcarnitine) of different acyl chain lengths (ranging from O-acetyl-(R)-carnitine to long-chain O-acyl-(R)-carnitines) with free carnitine ((R)-carnitine or L-carnitine) across the mitochondrial inner membrane, via a ping-pong mechanism (Probable) (PubMed:12892634, PubMed:18307102). Key player in the mitochondrial oxidation pathway, it translocates the fatty acids in the form of acylcarnitines into the mitochondr
Mitochondrion inner membrane
Carnitine-acylcarnitine translocase deficiency
A rare long-chain fatty acid oxidation disorder. Metabolic consequences include hypoketotic hypoglycemia under fasting conditions, hyperammonemia, elevated creatine kinase and transaminases, dicarboxylic aciduria, very low free carnitine and abnormal acylcarnitine profile with marked elevation of the long-chain acylcarnitines. Clinical features include neurologic abnormalities, cardiomyopathy, arrhythmias, skeletal muscle damage, liver dysfunction and episodes of life-threatening coma, which eventually lead to death. Most patients become symptomatic in the neonatal period with a rapidly progressive deterioration and a high mortality rate.
Sodium-ion dependent, high affinity carnitine transporter. Involved in the active cellular uptake of carnitine. Transports one sodium ion with one molecule of carnitine (PubMed:10454528, PubMed:10525100, PubMed:10966938, PubMed:17509700, PubMed:20722056, PubMed:33124720). Also transports organic cations such as tetraethylammonium (TEA) without the involvement of sodium. Relative uptake activity ratio of carnitine to TEA is 11.3 (PubMed:10454528, PubMed:10525100, PubMed:10966938). In intestinal e
Cell membraneApical cell membraneBasal cell membraneEndoplasmic reticulum
Systemic primary carnitine deficiency
Autosomal recessive disorder of fatty acid oxidation caused by defective carnitine transport. Present early in life with hypoketotic hypoglycemia and acute metabolic decompensation, or later in life with skeletal myopathy or cardiomyopathy.
Medicamentos e terapias
Mecanismo: Peroxisome proliferator-activated receptor agonist
Variantes genéticas (ClinVar)
1.492 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
5 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Alteração do ciclo de carnitina e transporte de carnitina
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Plasma Metabolites Associated with CKD Stage in Autosomal Dominant Tubulointerstitial Kidney Disease.
This is the first large-scale metabolomic study in genetically confirmed autosomal dominant tubulointerstitial kidney disease (ADTKD), providing a new resource for rare kidney diseases. ADTKD-UMOD and ADTKD-MUC1 are metabolically indistinguishable across stages, supporting the development of unified monitoring strategies. The plasma kynurenine-to-tryptophan ratio increases with CKD progression, supporting its use as a noninvasive marker of inflammation in ADTKD. Metabolomic profiling has not yet been performed in autosomal dominant tubulointerstitial kidney disease (ADTKD) due to UMOD or MUC1 mutations and could provide valuable insights into the pathophysiology of these conditions and identify the biomarkers of disease activity. Untargeted metabolomic analysis of plasma samples was performed on a cohort comprising 40 controls, 51 individuals with ADTKD-UMOD, and 49 individuals with ADTKD-MUC1 with CKD stages ranging from 1 to 4. Principal component analysis and hierarchical clustering revealed that the metabolic profiles of controls and ADTKD-UMOD and ADTKD-MUC1 patients with CKD stages 1 and 2 were similar. The metabolome was also similar between patients with ADTKD-UMOD and ADTKD-MUC1 at each stage of CKD. Compared with stage 2 CKD, stage 3 CKD was characterized by an increased kynurenine-to-tryptophan ratio, indicating activation of indoleamine 2,3-dioxygenase, an inflammation-induced and rate-limiting enzyme of tryptophan metabolism, and increased levels of pseudouridine, 3-indoxylsulfate, N-formylmethione, N-acetylated amino acids, acylcarnitines, and several other metabolites. In total, 121 metabolites were identified as significantly altered in patients in stage 4 compared with controls. Enrichment analysis of this set revealed that the most significant alterations were in the biosynthesis of arginine and branched-chain amino acids, carnitine synthesis, transfer RNA metabolism, tryptophan catabolism, urea cycle, metabolism of amino acids, glucose homeostasis, and solute carrier-mediated transmembrane transport. Patients with ADTKD-UMOD and ADTKD-MUC1 had similar metabolomic profiles across CKD stages. Although ADTKD is a tubulointerstitial kidney disease rather than glomerular, the effects on the metabolomic pathways appear comparable with those of other forms of CKD. The kynurenine-to-tryptophan ratio appears to be a promising biomarker of ADTKD progression and will require additional study.
Primary systemic carnitine deficiency: Phenotypic variability, diagnostic challenges, and long-term outcomes.
Primary systemic carnitine deficiency (CDSP) is a rare inherited metabolic disorder characterized by impaired carnitine transport due to mutations in the SLC22A5 gene, leading to impaired mitochondrial fatty acid oxidation. The aim of this retrospective, descriptive study was to investigate the clinical, biochemical, and molecular features of CDSP in Turkey, where the lack of a national expanded metabolic screening program contributes to delayed diagnosis and severe complications. The clinical, biochemical, and molecular profiles of 12 patients from eight families diagnosed between 2003 and 2025 were retrospectively analyzed. Data on family history, consanguinity, clinical manifestations-including cardiomyopathy, muscle weakness, neurological symptoms, and liver dysfunction-plasma free carnitine levels, and echocardiographic measurements were collected and analyzed. The majority of patients (92%) were from consanguineous families. Cardiomyopathy was the most common clinical feature (75%), followed by muscle weakness (50%), neurological symptoms (42%), and liver dysfunction (33%). A novel SLC22A5 variant (c.125T>C; p.Leu42Pro) was identified that expands the known genetic spectrum of CDSP. Oral carnitine supplementation significantly increased plasma free carnitine levels (p = 0.01) and improved long-term interventricular septal thickness Z-scores (p = 0.045). In addition, cholestasis was observed in two patients, suggesting an expanded disease phenotype. These results emphasize the crucial role of early detection and family screening in the prevention of life-threatening complications associated with CDSP. Long-term carnitine therapy improves metabolic and cardiac outcomes, underscoring the need for early intervention and inclusion of CDSP in national newborn screening programs.
PNPLA3 I148M variant links to adverse metabolic traits in MASLD during fasting and feeding.
The PNPLA3 rs738409 polymorphism is the most abundant genetic risk factor associated with progression of metabolic dysfunction-associated steatotic liver disease (MASLD) to steatohepatitis (MASH) and fibrosis. Although fasting and feeding affect PNPLA3 expression, molecular insights into the pathophysiological influence remain scarce. We analyzed 353 serum samples of patients with MASLD from two German tertiary centers using nuclear magnetic resonance (NMR)-proteometabolomics. Patients were stratified by PNPLA3 rs738409 C>G genotype: 'CC', 'CG', and 'GG'. Metabolites, lipoproteins, and glycoproteins were assessed based on fasting status. PNPLA3 GG displayed a distinct metabolic profile, with notable alterations between fasting and non-fasting states. During the latter, GG carriers showed lower levels of VLDL-1, reflecting impaired triglyceride (TG) efflux from hepatocytes. Following an overnight fast, GG carriers exhibited higher tricarboxylic acid cycle metabolites and ketone bodies, overall indicating increased β-oxidation likely attributed to lower PNPLA3 expression, facilitating unrestricted adipose triglyceride lipase activity and consecutive increased hepatic TG secretion. In addition, the ketogenic amino acid lysine, critical for mitochondrial carnitine transport, was significantly reduced (GG 0.14 ± 0.09 mM vs. CC 0.18 ± 0.08 mM, q = 0.015). Consistently, TGs were enriched in LDL and HDL particles, and an increased number of intermediate-density lipoproteins emerged as a distinct marker in fasted GG carriers (GG 202.9 ± 68.2 mg/dl vs. CC 160.8 ± 65.6 mg/dl, q = 0.007). These metabolic changes were enhanced in patients with type 2 diabetes mellitus and/or obesity. Our findings suggest a dichotomous pattern of increased hepatic lipid storage during feeding and excessive lipid oxidation during fasting, which exceeds metabolic capacity, inducing cellular toxicity in PNPLA3 GG carriers. This interplay fuels a detrimental fasting/non-fasting cycle, thus pointing to the need for preventive strategies. The PNPLA3 rs738409 (p.I148M) polymorphism is the most prevalent genetic risk factor for metabolic dysfunction-associated steatotic liver disease progression and is influenced by fasting and feeding cycles. However, the pathophysiological consequences of this regulation remain poorly understood. Nuclear magnetic resonance-proteometabolomics reveals a distinct signature in homozygous PNPLA3 GG carriers that changes significantly with fasting status, providing important implications for diagnosis and preventive strategies.
Development and validation of a carnitine cycle and transport disorders (CCD) panel: an ONT-compatible multi-gene diagnostic kit for newborn and selective screening.
Carnitine transport and cycle disorders (CCD) are a group of metabolic disorders characterized by either carnitine depletion or dysfunction in the carnitine cycle, a critical process for the transport of fatty acids into the mitochondria and their subsequent β-oxidation. Clinically, CCD can manifest with a wide range of symptoms, including hypoketotic hypoglycemia, which may be accompanied by signs of liver dysfunction, hepatic steatosis, myopathy and cardiomyopathy. Biochemical diagnosis typically involves measuring carnitine and acylcarnitine levels in blood, alongside organic acid profiling in urine. However, due to phenotypic overlaps with other metabolic disorders, precise molecular diagnosis is essential for accurate disease classification and subtype determination. The present study aimed to develop and clinically validate a novel CCD panel, specifically designed for Oxford Nanopore Technologies (ONT) platform compatibility. The panel targeted four key CCD related genes (CPT-1, CPT-2, SLC22A5 and SLC25A20). An amplification-based library preparation method pooling 21 primers specific to the CCD-related genes into two tubes was optimized. The panel was then applied to screen 20 patients previously diagnosed with CCD via second-generation sequencing platform. Comparative analysis of results from both platforms revealed a 100% concordance in detecting pathogenic, likely pathogenic, and variants of unknown significance associated with CCD. In silico analysis was also performed to predict the pathogenic potential of the variants of unknown significance. Here we report the development and clinical validation of a multi-gene diagnostic panel for ONT platform. The results demonstrated the feasibility of ONT-based genetic testing for CCD and set the stage for the development of similar diagnostic panels for other genetic disorders, offering a streamlined and putatively cost-effective alternative to current sequencing methodologies.
ndufs2-/- zebrafish have impaired survival, neuromuscular activity, morphology, and one-carbon metabolism treatable with folic acid.
Mitochondrial complex I (CI) deficiency represents a common biochemical pathophysiology underlying Leigh syndrome spectrum (LSS), manifesting with progressive multi-system dysfunction, lactic acidemia, and early mortality. To facilitate mechanistic studies and rigorous screening of therapeutic candidates for CI deficient LSS, we used CRISPR/Cas9 to generate an ndufs2 -/- 16 bp deletion zebrafish strain. ndufs2 -/- larvae exhibit markedly reduced survival, severe neuromuscular dysfunction including impaired swimming capacity, multiple morphologic malformations, reduced growth, hepatomegaly, uninflated swim bladder, yolk retention, small intestines, and small eyes and pupils with abnormal retinal ganglion cell layer. Transcriptome profiling of ndufs2 -/- larvae revealed dysregulation of the electron transport chain, TCA cycle, fatty acid beta-oxidation, and one-carbon metabolism. Similar transcriptomic profiles were observed in ndufs2 -/- missense mutant C. elegans (gas-1(fc21)) and two human CI-disease fibroblast cell lines stressed in galactose media. ndufs2 -/- zebrafish had 80% reduced CI enzyme activity. Unbiased metabolomic profiling showed increased lactate, TCA cycle intermediates, and acyl-carnitine species. One-carbon metabolism associated pathway alterations appear to contribute to CI disease pathophysiology, as folic acid treatment rescued the growth defect and hepatomegaly in ndufs2 -/- larvae. Overall, ndufs2 -/- zebrafish recapitulate severe CI deficiency, complex metabolic pathophysiology, and relevant LSS neuromuscular and survival phenotypes, enabling future translational studies of therapeutic candidates.
Publicações recentes
The mechanism of acetyl-L-carnitine on colorectal cancer and its metabolomic study.
Amino acid metabolism in diabetic kidney disease.
Primary systemic carnitine deficiency: Phenotypic variability, diagnostic challenges, and long-term outcomes.
Pitfalls in the diagnosis of carnitine palmitoyltransferase 1 deficiency.
The role of carnitine palmitoyl transferase 2 in the progression of salt-sensitive hypertension.
📚 EuropePMCmostrando 51
Plasma Metabolites Associated with CKD Stage in Autosomal Dominant Tubulointerstitial Kidney Disease.
Kidney360Primary systemic carnitine deficiency: Phenotypic variability, diagnostic challenges, and long-term outcomes.
Pediatrics international : official journal of the Japan Pediatric Societyndufs2-/- zebrafish have impaired survival, neuromuscular activity, morphology, and one-carbon metabolism treatable with folic acid.
bioRxiv : the preprint server for biologyPNPLA3 I148M variant links to adverse metabolic traits in MASLD during fasting and feeding.
JHEP reports : innovation in hepatologyThe global prevalence and genetic spectrum of primary carnitine deficiency.
BMC genomic dataDevelopment and validation of a carnitine cycle and transport disorders (CCD) panel: an ONT-compatible multi-gene diagnostic kit for newborn and selective screening.
Orphanet journal of rare diseasesMetabolic origin and significance of 3-methylglutaryl CoA.
Clinica chimica acta; international journal of clinical chemistryHyperornithinemia-Hyperammonemia-Homocitrullinuria Syndrome in Vietnamese Patients.
Medicina (Kaunas, Lithuania)Lipid storage myopathy associated with sertraline treatment is an acquired mitochondrial disorder with respiratory chain deficiency.
Acta neuropathologicaAllele-specific dysregulation of lipid and energy metabolism in early-stage hypertrophic cardiomyopathy.
Journal of molecular and cellular cardiology plusCoenzyme A biosynthesis: mechanisms of regulation, function and disease.
Nature metabolismPrevalence of inherited metabolic disorders among newborns in Zhuzhou, a southern city in China.
Frontiers in geneticsAluminum as a Possible Cause Toward Dyslipidemia.
Indian journal of occupational and environmental medicineUnderstanding the Pathogenesis of Cardiac Complications in Patients with Propionic Acidemia and Exploring Therapeutic Alternatives for Those Who Are Not Eligible or Are Waiting for Liver Transplantation.
MetabolitesMetabolomics-based safety evaluation of acute exposure to electronic cigarettes in mice.
The Science of the total environmentThe Reversible Carnitine Palmitoyltransferase 1 Inhibitor (Teglicar) Ameliorates the Neurodegenerative Phenotype in a Drosophila Huntington's Disease Model by Acting on the Expression of Carnitine-Related Genes.
Molecules (Basel, Switzerland)Anesthetic management of patients with carnitine deficiency or a defect of the fatty acid β-oxidation pathway: A narrative review.
MedicineMetabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma.
Acta pharmaceutica Sinica. BmiR-669a-5p promotes adipogenic differentiation and induces browning in preadipocytes.
AdipocyteProspective diagnosis of MT-ATP6-related mitochondrial disease by newborn screening.
Molecular genetics and metabolismAlterations in acylcarnitines, amines, and lipids inform about the mechanism of action of citalopram/escitalopram in major depression.
Translational psychiatryCombined primary carnitine deficiency with neonatal intrahepatic cholestasis caused by citrin deficiency in a Chinese newborn.
BMC pediatricsThe Discovery of Highly Potent THP Derivatives as OCTN2 Inhibitors: From Structure-Based Virtual Screening to In Vivo Biological Activity.
International journal of molecular sciencesPrimary carnitine deficiency in two sisters with intractable epilepsy and reversible metabolic cardiomyopathy: Two case reports.
Experimental and therapeutic medicineEnhanced fatty acid oxidation mediated by CPT1C promotes gastric cancer progression.
Journal of gastrointestinal oncologyClinical and biological characterization of 20 patients with TANGO2 deficiency indicates novel triggers of metabolic crises and no primary energetic defect.
Journal of inherited metabolic diseaseFriedreich Ataxia: current state-of-the-art, and future prospects for mitochondrial-focused therapies.
Translational research : the journal of laboratory and clinical medicineTherapeutic effect of N-carbamylglutamate in CPS1 deficiency.
Molecular genetics and metabolism reportsWeaning Alters Intestinal Gene Expression Involved in Nutrient Metabolism by Shaping Gut Microbiota in Pigs.
Frontiers in microbiologyDiseases Caused by Mutations in Mitochondrial Carrier Genes SLC25: A Review.
BiomoleculesFunctional changes of the liver in the absence of growth hormone (GH) action - Proteomic and metabolomic insights from a GH receptor deficient pig model.
Molecular metabolismFever, Fasting, and Rhabdomyolysis in an Adult Male.
Neurology IndiaA metabolomics-based molecular pathway analysis of how the sodium-glucose co-transporter-2 inhibitor dapagliflozin may slow kidney function decline in patients with diabetes.
Diabetes, obesity & metabolismPotential Therapeutic Role of Carnitine and Acetylcarnitine in Neurological Disorders.
Current pharmaceutical designNutritional ketosis improves exercise metabolism in patients with very long-chain acyl-CoA dehydrogenase deficiency.
Journal of inherited metabolic diseaseCarnitine Inborn Errors of Metabolism.
Molecules (Basel, Switzerland)xCT knockdown in human breast cancer cells delays onset of cancer-induced bone pain.
Molecular painLow-dose cadmium disrupts mitochondrial citric acid cycle and lipid metabolism in mouse lung.
Free radical biology & medicineThe SLC22 Transporter Family: A Paradigm for the Impact of Drug Transporters on Metabolic Pathways, Signaling, and Disease.
Annual review of pharmacology and toxicologyA novel homozygous SLC25A1 mutation with impaired mitochondrial complex V: Possible phenotypic expansion.
American journal of medical genetics. Part A[Clinical diagnosis and treatment of three cases with hyperornithinemia-hyperammonemia-homocitrullinuria syndrome].
Zhonghua er ke za zhi = Chinese journal of pediatricsPrimary Carnitine Deficiency and Newborn Screening for Disorders of the Carnitine Cycle.
Annals of nutrition & metabolism36th International Symposium on Intensive Care and Emergency Medicine : Brussels, Belgium. 15-18 March 2016.
Critical care (London, England)Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency.
International journal of molecular sciencesIncreased maternal consumption of methionine as its hydroxyl analog promoted neonatal intestinal growth without compromising maternal energy homeostasis.
Journal of animal science and biotechnologyThe odd-carbon medium-chain fatty triglyceride triheptanoin does not reduce hepatic steatosis.
Clinical nutrition (Edinburgh, Scotland)Exercise and Regulation of Lipid Metabolism.
Progress in molecular biology and translational scienceModification of Astrocyte Metabolism as an Approach to the Treatment of Epilepsy: Triheptanoin and Acetyl-L-Carnitine.
Neurochemical researchBiotin deprivation impairs mitochondrial structure and function and has implications for inherited metabolic disorders.
Molecular genetics and metabolismMitochondrial vulnerability and increased susceptibility to nutrient-induced cytotoxicity in fibroblasts from leigh syndrome French canadian patients.
PloS oneVitamin E in New-Generation Lipid Emulsions Protects Against Parenteral Nutrition-Associated Liver Disease in Parenteral Nutrition-Fed Preterm Pigs.
JPEN. Journal of parenteral and enteral nutritionAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Alteração do ciclo de carnitina e transporte de carnitina.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Alteração do ciclo de carnitina e transporte de carnitina
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Plasma Metabolites Associated with CKD Stage in Autosomal Dominant Tubulointerstitial Kidney Disease.
- Primary systemic carnitine deficiency: Phenotypic variability, diagnostic challenges, and long-term outcomes.Pediatrics international : official journal of the Japan Pediatric Society· 2025· PMID 41048097mais citado
- PNPLA3 I148M variant links to adverse metabolic traits in MASLD during fasting and feeding.
- Development and validation of a carnitine cycle and transport disorders (CCD) panel: an ONT-compatible multi-gene diagnostic kit for newborn and selective screening.
- ndufs2-/- zebrafish have impaired survival, neuromuscular activity, morphology, and one-carbon metabolism treatable with folic acid.
- The mechanism of acetyl-L-carnitine on colorectal cancer and its metabolomic study.
- Amino acid metabolism in diabetic kidney disease.
- Pitfalls in the diagnosis of carnitine palmitoyltransferase 1 deficiency.
- The role of carnitine palmitoyl transferase 2 in the progression of salt-sensitive hypertension.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:309130(Orphanet)
- MONDO:0017716(MONDO)
- GARD:21320(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55787303(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
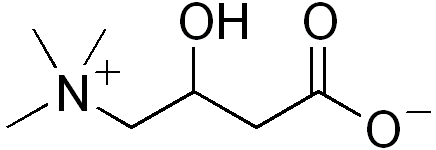
Alteração do ciclo de carnitina e transporte de carnitina
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos (literatura)
- fonte: Orphanet