A síndrome de Angelman (SA) é um distúrbio genético raro que afeta aproximadamente 1 em cada 15.000 indivíduos. A SA prejudica a função do sistema nervoso, produzindo sintomas como deficiência intelectual grave, deficiência no desenvolvimento, fala funcional limitada ou ausente, problemas de equilíbrio e movimento, convulsões, hiperatividade e problemas de sono. Os sintomas físicos incluem uma cabeça pequena e uma aparência facial específica. Além disso, os indivíduos afetados geralmente possuem uma personalidade alegre. A síndrome de Angelman envolve genes que também foram associados a 1–2% dos casos de transtorno do espectro autista.
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome Angelman por mutação pontual é uma condição genética rara que afeta o desenvolvimento neurológico, geralmente identificada na primeira infância. Ela é causada por alterações específicas no gene UBE3A, que codifica a enzima ubiquitina-proteína ligase E3A, essencial para o funcionamento normal do sistema nervoso. A condição se manifesta por uma combinação de características físicas, neurológicas e comportamentais, como atraso no desenvolvimento, convulsões e um padrão de comportamento alegre e sorridente.[1][3]
Sinais e sintomas
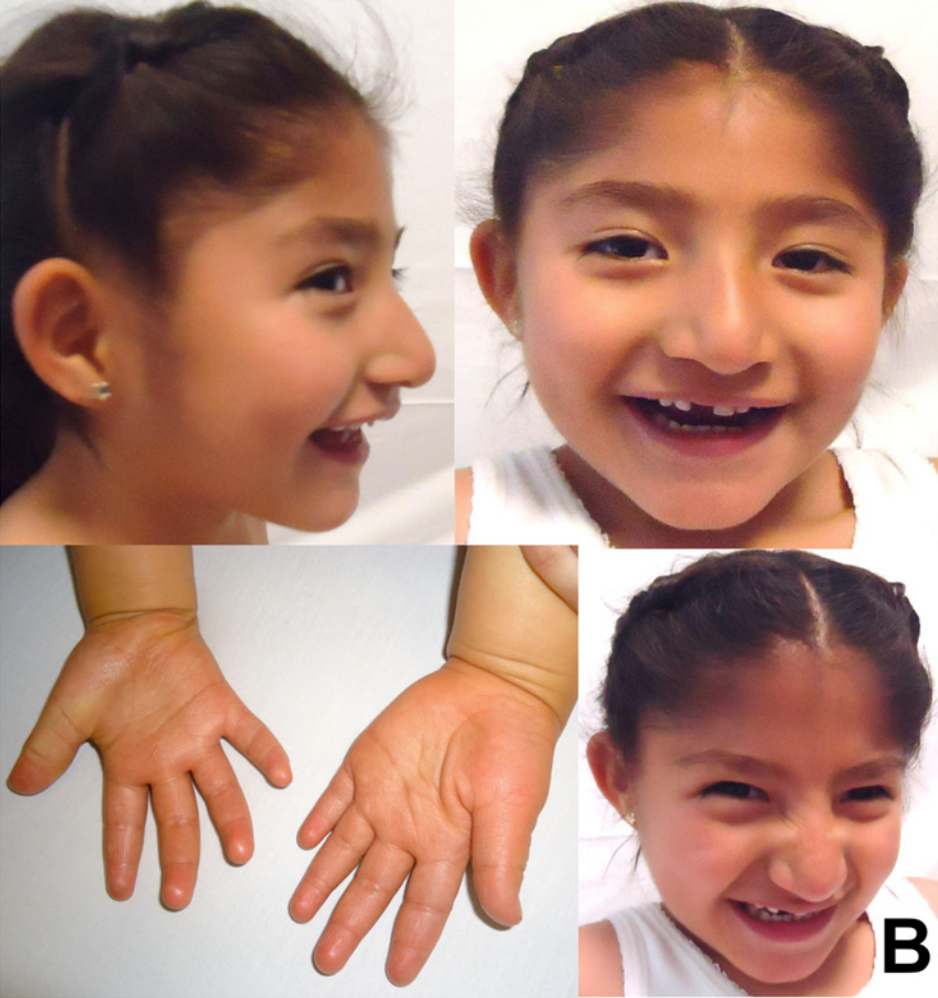
Os sinais e sintomas da Síndrome Angelman por mutação pontual incluem uma ampla variedade de manifestações. Entre os fenótipos mais comuns estão: estrabismo, hiperreflexia dos membros inferiores, occipital plano (achatamento da parte de trás da cabeça), microcefalia leve, prognatismo mandibular (projeção da mandíbula), boca larga, língua protusa, hipopigmentação da pele, cabelo e íris, e comportamento alimentar anormal com sucção pobre e dificuldades alimentares. Também são frequentes intolerância ao calor, cessação do crescimento da cabeça, obesidade, ataxia (falta de coordenação motora), desequilíbrio da marcha, marcha de base alargada, convulsões, anormalidades no EEG, deficiência intelectual leve, anormalidades da fala ou vocalização, e o característico comportamento alegre com flapping de mãos recorrente.[1][3]
Causas genéticas
A Síndrome Angelman por mutação pontual é causada por mutações no gene UBE3A (Ubiquitin-protein ligase E3A), localizado no cromossomo 15. Esse gene é responsável pela produção de uma enzima que participa da degradação de proteínas dentro das células, processo crucial para o desenvolvimento e funcionamento do cérebro. A herança genética e a prevalência exata dessa forma específica da síndrome não estão completamente estabelecidas nos dados disponíveis.[1][4]
Diagnóstico
O diagnóstico da Síndrome Angelman por mutação pontual é baseado na avaliação clínica dos sinais e sintomas característicos, confirmado por testes genéticos. Os procedimentos disponíveis no SUS incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); dosagem de alfa-fetoproteína; e atendimento em reabilitação para doenças raras. Atualmente, há 336 testes genéticos registrados e 761 variantes documentadas no ClinVar relacionadas a essa condição.[1][4]
Tratamento e manejo
O manejo da Síndrome Angelman por mutação pontual é multidisciplinar e focado no controle dos sintomas e na melhora da qualidade de vida. Inclui acompanhamento neurológico para convulsões, fisioterapia para ataxia e distúrbios da marcha, terapia ocupacional e fonoaudiologia para dificuldades alimentares e de fala, além de suporte educacional para deficiência intelectual. O nível de cobertura pelo SUS é classificado como cobertura mínima, e os procedimentos disponíveis incluem atendimento em reabilitação para doenças raras. Não há medicamentos específicos listados nos dados oficiais para essa condição.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Síndrome Angelman por mutação pontual varia conforme a gravidade dos sintomas e o acesso a intervenções precoces. Embora a deficiência intelectual e os desafios motores e de fala persistam ao longo da vida, muitos indivíduos apresentam um comportamento alegre e interagem socialmente. O acompanhamento regular com equipe multidisciplinar e o suporte familiar são fundamentais para promover a melhor qualidade de vida possível.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
A síndrome de Angelman (SA) é um distúrbio genético raro que afeta aproximadamente 1 em cada 15.000 indivíduos. A SA prejudica a função do sistema nervoso, produzindo sintomas como deficiência intelectual grave, deficiência no desenvolvimento, fala funcional limitada ou ausente, problemas de equilíbrio e movimento, convulsões, hiperatividade e problemas de sono. Os sintomas físicos incluem uma cabeça pequena e uma aparência facial específica. Além disso, os indivíduos afetados geralmente possuem uma personalidade alegre. A síndrome de Angelman envolve genes que também foram associados a 1–2% dos casos de transtorno do espectro autista.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome Angelman por mutação pontual é uma condição genética rara que afeta o desenvolvimento neurológico, geralmente identificada na primeira infância. Ela é causada por alterações específicas no gene UBE3A, que codifica a enzima ubiquitina-proteína ligase E3A, essencial para o funcionamento normal do sistema nervoso. A condição se manifesta por uma combinação de características físicas, neurológicas e comportamentais, como atraso no desenvolvimento, convulsões e um padrão de comportamento alegre e sorridente.[1][3]
Sinais e sintomas
Os sinais e sintomas da Síndrome Angelman por mutação pontual incluem uma ampla variedade de manifestações. Entre os fenótipos mais comuns estão: estrabismo, hiperreflexia dos membros inferiores, occipital plano (achatamento da parte de trás da cabeça), microcefalia leve, prognatismo mandibular (projeção da mandíbula), boca larga, língua protusa, hipopigmentação da pele, cabelo e íris, e comportamento alimentar anormal com sucção pobre e dificuldades alimentares. Também são frequentes intolerância ao calor, cessação do crescimento da cabeça, obesidade, ataxia (falta de coordenação motora), desequilíbrio da marcha, marcha de base alargada, convulsões, anormalidades no EEG, deficiência intelectual leve, anormalidades da fala ou vocalização, e o característico comportamento alegre com flapping de mãos recorrente.[1][3]
Causas genéticas
A Síndrome Angelman por mutação pontual é causada por mutações no gene UBE3A (Ubiquitin-protein ligase E3A), localizado no cromossomo 15. Esse gene é responsável pela produção de uma enzima que participa da degradação de proteínas dentro das células, processo crucial para o desenvolvimento e funcionamento do cérebro. A herança genética e a prevalência exata dessa forma específica da síndrome não estão completamente estabelecidas nos dados disponíveis.[1][4]
Diagnóstico
O diagnóstico da Síndrome Angelman por mutação pontual é baseado na avaliação clínica dos sinais e sintomas característicos, confirmado por testes genéticos. Os procedimentos disponíveis no SUS incluem: cariótipo com bandas G, Q ou R; pesquisa de microdeleções/microduplicações por FISH; sequenciamento completo do exoma (WES); dosagem de alfa-fetoproteína; e atendimento em reabilitação para doenças raras. Atualmente, há 336 testes genéticos registrados e 761 variantes documentadas no ClinVar relacionadas a essa condição.[1][4]
Tratamento e manejo
O manejo da Síndrome Angelman por mutação pontual é multidisciplinar e focado no controle dos sintomas e na melhora da qualidade de vida. Inclui acompanhamento neurológico para convulsões, fisioterapia para ataxia e distúrbios da marcha, terapia ocupacional e fonoaudiologia para dificuldades alimentares e de fala, além de suporte educacional para deficiência intelectual. O nível de cobertura pelo SUS é classificado como cobertura mínima, e os procedimentos disponíveis incluem atendimento em reabilitação para doenças raras. Não há medicamentos específicos listados nos dados oficiais para essa condição.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Síndrome Angelman por mutação pontual varia conforme a gravidade dos sintomas e o acesso a intervenções precoces. Embora a deficiência intelectual e os desafios motores e de fala persistam ao longo da vida, muitos indivíduos apresentam um comportamento alegre e interagem socialmente. O acompanhamento regular com equipe multidisciplinar e o suporte familiar são fundamentais para promover a melhor qualidade de vida possível.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 16 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 34 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição.
E3 ubiquitin-protein ligase which accepts ubiquitin from an E2 ubiquitin-conjugating enzyme in the form of a thioester and transfers it to its substrates (PubMed:10373495, PubMed:16772533, PubMed:19204938, PubMed:19233847, PubMed:19325566, PubMed:19591933, PubMed:22645313, PubMed:24273172, PubMed:24728990, PubMed:30020076). Several substrates have been identified including the BMAL1, ARC, LAMTOR1, RAD23A and RAD23B, MCM7 (which is involved in DNA replication), annexin A1, the PML tumor suppresso
CytoplasmNucleus
Angelman syndrome
A neurodevelopmental disorder characterized by severe motor and intellectual retardation, ataxia, frequent jerky limb movements and flapping of the arms and hands, hypotonia, seizures, absence of speech, frequent smiling and episodes of paroxysmal laughter, open-mouthed expression revealing the tongue.
Variantes genéticas (ClinVar)
761 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Angelman por mutação pontual
Centros de Referência SUS
24 centros habilitados pelo SUS para Síndrome Angelman por mutação pontual
Centros para Síndrome Angelman por mutação pontual
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Mutation of ube3a causes developmental abnormalities and autism-like molecular and behavioral alterations in zebrafish.
Mutations in the UBE3A gene are responsible for neurodevelopmental disorders (NDDs), including Angelman syndrome (AS), which is characterized by developmental delays, impaired motor coordination, and cognitive disabilities. In recent years, UBE3A mutations have also been linked to autism spectrum disorders (ASD), due to their significant role in synaptic plasticity and cognitive function. Although substantial research has utilized mammalian models, the zebrafish (Danio rerio) provides unique opportunities to investigate gene functions owing to their transparent embryos, rapid development, and suitability for large-scale genetic and behavioral studies. In this study, we characterized a zebrafish model harboring a point mutation (T > A) in exon 3 of the zebrafish ube3a gene, which induces a stop codon resulting in a truncated protein. We performed comprehensive developmental, behavioral, and molecular analyses to investigate the impact of Ube3a dysfunction at both larval and adult stages. We observed alterations in embryonic development, significant locomotor deficits, including stereotypic movements, and reduced social preference and aggressiveness. Furthermore, RNA sequencing analysis of both larvae and adults revealed dysregulation in chromatin, nucleosome, protein-DNA, and primary cilia-related genes. Our findings provide a functional characterization of the ube3a mutation in zebrafish at both larval and adult stages. This zebrafish model offers new insights into the roles of UBE3A in neurodevelopment and behavior, expanding our understanding of its dysfunction in NDDs.
From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research.
Angelman syndrome is a rare neurodevelopmental disorder caused by mutations affecting the chromosomal 15q11-13 region, either by contiguous gene deletions, imprinting defects, uniparental disomy, or mutations in the UBE3A gene itself. Phenotypic abnormalities are driven primarily, but not exclusively (especially in 15q11-13 deletion cases) by loss of expression of the maternally inherited UBE3A gene expression. The disorder was first described in 1965 by the English pediatrician Harry Angelman. Since that first description of three children with Angelman syndrome, there has been extensive research into the genetic, molecular and phenotypic aspects of the disorder. In the last decade, this has resulted in over 100 publications per year. Collectively, this research has led the field to a pivotal point in which restoring UBE3A function by genetic therapies is currently explored in several clinical trials. In this study, we employed a bibliometric approach to review and visualize the development of Angelman syndrome research over the last 50 years. We look into different parameters shaping the progress of the Angelman syndrome research field, including source of funding, publishing journals and international collaborations between research groups. Using a network approach, we map the focus of the research field and how that shifted over time. This overview helps understand the shift of research focus in the field and can provide a comprehensive handbook of Angelman syndrome research development.
The role of whole exome sequencing in the UBE3A point mutation of Angelman Syndrome: A case report.
Angelman Syndrome (AS) is a rare disorder with a relatively well-defined phenotype caused by lack of expression of the maternally inherited ubiquitin-protein ligase E3A (UBE3A) gene in the brain. This article describes the role of genetic testing using whole-exome sequencing (WES) in detecting rare AS variants, a point mutation in the UBE3A gene. We describe a rarely reported clinical presentation of AS in a two year and ten months old girl with severe developmental delay, movement and balance disorder, frequent smiling, apparent happy demeanor, speech impairment, absence of seizure, lack of sleep, and abnormal food-related behavior. Physical examination showed microcephaly, with facial characteristics of AS, ataxia gait, and truncal hypotonia. The electroencephalogram showed medium amplitude rhythmic 2-3c/s. Brain Magnetic Resonance Imaging revealed microcephaly, corpus callosum dysgenesis, and heterotopia grey matter on the bilateral lateral ventricle. WES was conducted to search pathogenic variants and showed a heterozygous mutation in exon 9 of the UBE3A gene, c.1513C > T (p.Arg505Ter). Angelman syndrome is a neurodevelopmental disorder that has several underlying genetic etiologies. WES could detect a rare variant of Angelman syndrome, identified as the point mutation of the UBE3A gene, which cannot be seen with other modalities.
Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants.
Genetic aberrations of the UBE3A gene encoding the E3 ubiquitin ligase E6AP underlie the development of Angelman syndrome (AS). Approximately 10% of AS individuals harbor UBE3A genes with point mutations, frequently resulting in the expression of full-length E6AP variants with defective E3 activity. Since E6AP exists in two states, an inactive and an active one, we hypothesized that distinct small molecules can stabilize the active state and that such molecules may rescue the E3 activity of AS-derived E6AP variants. Therefore, we established an assay that allows identifying modulators of E6AP in a high-throughput format. We identified several compounds that not only stimulate wild-type E6AP but also rescue the E3 activity of certain E6AP variants. Moreover, by chemical cross-linking coupled to mass spectrometry we provide evidence that the compounds stabilize an active conformation of E6AP. Thus, these compounds represent potential lead structures for the design of drugs for AS treatment.
An Angelman syndrome substitution in the HECT E3 ubiquitin ligase C-terminal Lobe of E6AP affects protein stability and activity.
Angelman syndrome (AS) is a rare neurodevelopmental disorder characterized by speech impairment, intellectual disability, ataxia, and epilepsy. AS is caused by mutations in the maternal copy of UBE3A located on chromosome 15q11-13. UBE3A codes for E6AP (E6 Associated Protein), a prominent member of the HECT (Homologous to E6AP C-Terminus) E3 ubiquitin ligase family. E6AP catalyzes the posttranslational attachment of ubiquitin via its HECT domain onto various intracellular target proteins to regulate DNA repair and cell cycle progression. The HECT domain consists of an N-lobe, required for E2~ubiquitin recruitment, while the C-lobe contains the conserved catalytic cysteine required for ubiquitin transfer. Previous genetic studies of AS patients have identified point mutations in UBE3A that result in amino acid substitutions or premature termination during translation. An AS transversion mutation (codon change from ATA to AAA) within the region of the gene that codes for the catalytic HECT domain of E6AP has been annotated (I827K), but the molecular basis for this loss of function substitution remained elusive. Here, we demonstrate that the I827K substitution destabilizes the 3D fold causing protein aggregation of the C-terminal lobe of E6AP using a combination of spectropolarimetry and nuclear magnetic resonance (NMR) spectroscopy. Our fluorescent ubiquitin activity assays with E6AP-I827K show decreased ubiquitin thiolester formation and ubiquitin discharge. Using 3D models in combination with our biochemical and biophysical results, we rationalize why the I827K disrupts E6AP-dependent ubiquitylation. This work provides new insight into the E6AP mechanism and how its malfunction can be linked to the AS phenotype.
Publicações recentes
Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants.
A proteasomal partner goes missing in Angelman syndrome.
Angelman syndrome-associated point mutations in the Zn(2+)-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome.
Psychiatric features in children with genetic syndromes: toward functional phenotypes.
Angelman syndrome due to a novel splicing mutation of the UBE3A gene.
📚 EuropePMCmostrando 11
Mutation of ube3a causes developmental abnormalities and autism-like molecular and behavioral alterations in zebrafish.
Brain research bulletinFrom first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research.
Human geneticsThe role of whole exome sequencing in the UBE3A point mutation of Angelman Syndrome: A case report.
Annals of medicine and surgery (2012)Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants.
Cell chemical biologyAn Angelman syndrome substitution in the HECT E3 ubiquitin ligase C-terminal Lobe of E6AP affects protein stability and activity.
PloS oneAngelman syndrome: a journey through the brain.
The FEBS journalGene mutations in paediatric epilepsies cause NMDA-pathy, and phasic and tonic GABA-pathy.
Developmental medicine and child neurologyA proteasomal partner goes missing in Angelman syndrome.
The Journal of biological chemistryAngelman syndrome-associated point mutations in the Zn2+-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome.
The Journal of biological chemistry[Clinical and genetic analysis of two unrelated patients with Angelman syndrome and novel UBE3A mutations].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsAngelman syndrome and isovaleric acidemia: What is the link?
Molecular genetics and metabolism reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Angelman por mutação pontual.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Angelman por mutação pontual
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Mutation of ube3a causes developmental abnormalities and autism-like molecular and behavioral alterations in zebrafish.
- From first report to clinical trials: a bibliometric overview and visualization of the development of Angelman syndrome research.
- The role of whole exome sequencing in the UBE3A point mutation of Angelman Syndrome: A case report.
- Identification of Small-Molecule Activators of the Ubiquitin Ligase E6AP/UBE3A and Angelman Syndrome-Derived E6AP/UBE3A Variants.
- An Angelman syndrome substitution in the HECT E3 ubiquitin ligase C-terminal Lobe of E6AP affects protein stability and activity.
- A proteasomal partner goes missing in Angelman syndrome.
- Angelman syndrome-associated point mutations in the Zn(2+)-binding N-terminal (AZUL) domain of UBE3A ubiquitin ligase inhibit binding to the proteasome.
- Psychiatric features in children with genetic syndromes: toward functional phenotypes.
- Angelman syndrome due to a novel splicing mutation of the UBE3A gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:411511(Orphanet)
- MONDO:0018461(MONDO)
- GARD:21732(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55788100(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Angelman por mutação pontual
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata