COG7-CDG é uma doença congênita (de nascença) que afeta a forma como o corpo processa certas moléculas essenciais, como açúcares e proteínas. Ela se manifesta com características físicas incomuns, problemas nos ossos, fraqueza muscular, aumento do fígado e do baço, pele e olhos amarelados (icterícia), coração fraco, infecções frequentes e epilepsia. Até o momento, apenas dois casos foram descritos, ambos em bebês que faleceram nos primeiros três meses de vida. Essa síndrome é causada por uma alteração (mutação) no gene COG-7 (localizado no cromossomo 16), que é responsável por uma parte de uma estrutura celular importante chamada Complexo de Golgi.
Introdução
O que você precisa saber de cara
COG7-CDG é uma doença congênita (de nascença) que afeta a forma como o corpo processa certas moléculas essenciais, como açúcares e proteínas. Ela se manifesta com características físicas incomuns, problemas nos ossos, fraqueza muscular, aumento do fígado e do baço, pele e olhos amarelados (icterícia), coração fraco, infecções frequentes e epilepsia. Até o momento, apenas dois casos foram descritos, ambos em bebês que faleceram nos primeiros três meses de vida. Essa síndrome é causada por uma alteração (mutação) no gene COG-7 (localizado no cromossomo 16), que é responsável por uma parte de uma estrutura celular importante chamada Complexo de Golgi.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 23 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 73 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
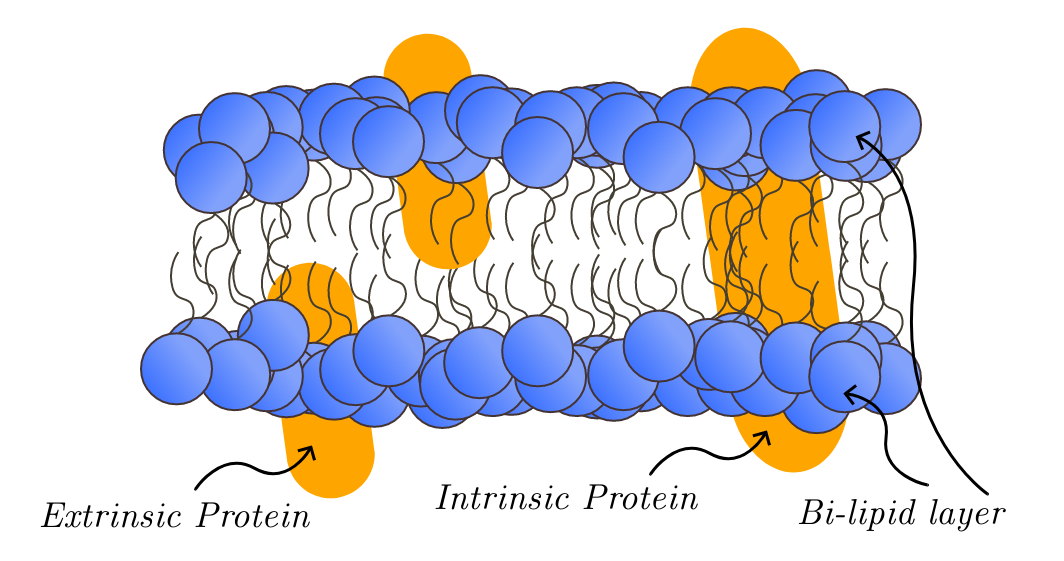
fontes oficiaisRequired for normal Golgi function
Golgi apparatus membrane
Congenital disorder of glycosylation 2E
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions.
Variantes genéticas (ClinVar)
75 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — COG7-CDG
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects.
The conserved oligomeric Golgi (COG) complex controls membrane trafficking and ensures Golgi homeostasis by orchestrating retrograde vesicle trafficking within the Golgi. Human COG defects lead to severe multisystemic diseases known as COG-congenital disorders of glycosylation (COG-CDG). To gain better understanding of COG-CDGs, we compared COG knockout cells with cells deficient to 2 key enzymes, Alpha-1,3-mannosyl-glycoprotein 2-beta-N-acetylglucosaminyltransferase and uridine diphosphate-glucose 4-epimerase (GALE), which contribute to proper N- and O-glycosylation. While all knockout cells share similar defects in glycosylation, these defects only account for a small fraction of observed COG knockout phenotypes. Glycosylation deficiencies were not associated with the fragmented Golgi, abnormal endolysosomes, defective sorting and secretion or delayed retrograde trafficking, indicating that these phenotypes are probably not due to hypoglycosylation, but to other specific interactions or roles of the COG complex. Importantly, these COG deficiency specific phenotypes were also apparent in COG7-CDG patient fibroblasts, proving the human disease relevance of our CRISPR knockout findings. The knowledge gained from this study has important implications, both for understanding the physiological role of COG complex in Golgi homeostasis in eukaryotic cells, and for better understanding human diseases associated with COG/Golgi impairment.
COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes.
Congenital disorders of glycosylation (CDG) comprise a family of human multisystemic diseases caused by recessive mutations in genes required for protein N-glycosylation. More than 100 distinct forms of CDGs have been identified and most of them cause severe neurological impairment. The Conserved Oligomeric Golgi (COG) complex mediates tethering of vesicles carrying glycosylation enzymes across the Golgi cisternae. Mutations affecting human COG1, COG2 and COG4-COG8 cause monogenic forms of inherited, autosomal recessive CDGs. We have generated a Drosophila COG7-CDG model that closely parallels the pathological characteristics of COG7-CDG patients, including pronounced neuromotor defects associated with altered N-glycome profiles. Consistent with these alterations, larval neuromuscular junctions of Cog7 mutants exhibit a significant reduction in bouton numbers. We demonstrate that the COG complex cooperates with Rab1 and Golgi phosphoprotein 3 to regulate Golgi trafficking and that overexpression of Rab1 can rescue the cytokinesis and locomotor defects associated with loss of Cog7. Our results suggest that the Drosophila COG7-CDG model can be used to test novel potential therapeutic strategies by modulating trafficking pathways.
Publicações recentes
More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects.
COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes.
Congenital disorders of glycosylation with emphasis on cerebellar involvement.
Wrinkled skin and fat pads in patients with ALG8-CDG: revisiting skin manifestations in congenital disorders of glycosylation.
COG5-CDG: expanding the clinical spectrum.
📚 EuropePMCmostrando 2
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para COG7-CDG.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para COG7-CDG
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- More than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects.
- COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes.
- Congenital disorders of glycosylation with emphasis on cerebellar involvement.
- Wrinkled skin and fat pads in patients with ALG8-CDG: revisiting skin manifestations in congenital disorders of glycosylation.
- COG5-CDG: expanding the clinical spectrum.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:79333(Orphanet)
- OMIM OMIM:608779(OMIM)
- MONDO:0012118(MONDO)
- GARD:9842(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q60195111(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
COG7-CDG
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata