A Hawkinsinúria é um erro congênito (presente desde o nascimento) no metabolismo da tirosina, um aminoácido. Ela é caracterizada por: dificuldade para ganhar peso e crescer, uma acidez persistente no sangue (acidose metabólica), cabelo fino e ralo, e pela eliminação, na urina, de uma substância química incomum chamada hawkinsina (que é um tipo de aminoácido cíclico).
Introdução
O que você precisa saber de cara
A Hawkinsinúria é um erro congênito (presente desde o nascimento) no metabolismo da tirosina, um aminoácido. Ela é caracterizada por: dificuldade para ganhar peso e crescer, uma acidez persistente no sangue (acidose metabólica), cabelo fino e ralo, e pela eliminação, na urina, de uma substância química incomum chamada hawkinsina (que é um tipo de aminoácido cíclico).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 9 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 17 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Curadoria gene-doença
fontes oficiaisCatalyzes the conversion of 4-hydroxyphenylpyruvic acid to homogentisic acid, one of the steps in tyrosine catabolism
CytoplasmEndoplasmic reticulum membraneGolgi apparatus membrane
Tyrosinemia 3
An autosomal recessive inborn error of metabolism characterized by high levels of tyrosine in the blood, massive excretion of its derivatives into urine, seizures and mild intellectual disability.
Variantes genéticas (ClinVar)
91 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 370 variantes classificadas pelo ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Hawkinsinúria
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Challenges in diagnosing hawkinsinuria in adulthood: 2 cases from a single family.
Hawkinsinuria, caused by an autosomal dominant gain-of-function variant of 4-hydroxyphenylpyruvate dioxygenase resulting in accumulation of 2-L-cystein-S-yl-1,4-dihydroxy-cyclohex-5-en-1-yl acetic acid (hawkinsin) and tyrosine, typically presents in the neonatal period. Here we report the case of a female adult patient in her early 20s presenting with childhood developmental delay and dyspraxia. She was initially referred to neurology, where baseline imaging and biochemistry were unremarkable. She was subsequently investigated for metabolic disorders, and it was found that plasma organic acids and amino acids were indicative of hawkinsinuria. Furthermore, her mother, who was asymptomatic, was also diagnosed with hawkinsinuria following family screening. Management was conservative, with regular monitoring of tyrosine and phenylalanine levels. Dietary restriction may be considered if tyrosine is elevated or patients become symptomatic. To our knowledge, this is the first reported case of hawkinsinuria presenting symptomatically in an adult patient and the second case of an asymptomatic adult being diagnosed from genetic testing.
Molecular and Evolution In Silico Studies Unlock the h4-HPPD C-Terminal Tail Gating Mechanism.
The enzyme 4-hydroxyphenylpyruvate dioxygenase (4-HPPD) is involved in the catabolism of the amino acid tyrosine in organisms such as bacteria, plants, and animals. It catalyzes the conversion of 4-hydroxyphenylpyruvate to a homogenisate in the presence of molecular oxygen and Fe(II) as a cofactor. This enzyme represents a key step in the biosynthesis of important compounds, and its activity deficiency leads to severe, rare autosomal recessive disorders, like tyrosinemia type III and hawkinsinuria, for which no cure is currently available. The 4-HPPD C-terminal tail plays a crucial role in the enzyme catalysis/gating mechanism, ensuring the integrity of the active site for catalysis through fine regulation of the C-terminal tail conformation. However, despite growing interest in the 4-HPPD catalytic mechanism and structure, the gating mechanism remains unclear. Furthermore, the absence of the whole 3D structure makes the bioinformatic approach the only possible study to define the enzyme structure/molecular mechanism. Here, wild-type 4-HPPD and its mutants were deeply dissected by applying a comprehensive bioinformatics/evolution study, and we showed for the first time the entire molecular mechanism and regulation of the enzyme gating process, proposing the full-length 3D structure of human 4-HPPD and two novel key residues involved in the 4-HPPD C-terminal tail conformational change.
A sportomics soccer investigation unveils an exercise-induced shift in tyrosine metabolism leading to hawkinsinuria.
Tyrosine metabolism has an intense role in the synthesis of neurotransmitters. Our study used an untargeted, sportomics-based analysis of urine samples to investigate changes in metabolism during a soccer match in 30 male junior professional soccer players. Samples were collected before and after the match and analyzed using liquid chromatography and mass spectrometry. Results showed significant changes in tyrosine metabolism. Exercise caused a downregulation of the homogentisate metabolites 4-maleylacetoacetate and succinylacetone to 20% (p = 4.69E-5) and 16% (p = 4.25E-14), respectively. 4-Hydroxyphenylpyruvate, a homogentisate precursor, was found to be upregulated by 26% (p = 7.20E-3). The concentration of hawkinsin and its metabolite 4-hydroxycyclohexyl acetate increased ~six-fold (p = 1.49E-6 and p = 9.81E-6, respectively). Different DOPA metabolism pathways were also affected by exercise. DOPA and dopaquinone increased four-to six-fold (p = 5.62E-14 and p = 4.98E-13, respectively). 3-Methoxytyrosine, indole-5,6-quinone, and melanin were downregulated from 1 to 25%, as were dopamine and tyramine (decreasing to up to 5% or 80%; p= 5.62E-14 and p = 2.47E-2, respectively). Blood TCO2 decreased as well as urinary glutathione and glutamate (40% and 10% respectively) associated with a two-fold increase in pyroglutamate. Our study found unexpected similarities between exercise-induced changes in metabolism and the inherited disorder Hawkinsinuria, suggesting a possible transient condition called exercise-induced hawkinsinuria (EIh). Additionally, our research suggests changes in DOPA pathways may be involved. Our findings suggest that soccer exercise could be used as a model to search for potential countermeasures in Hawkinsinuria and other tyrosine metabolism disorders.
Variant analysis of HPD genes from two families showing elevated tyrosine upon newborn screening by tandem mass spectrometry (MS/MS).
Background Alterations in the structure and activity of 4-hydroxyphenylpyruvate dioxygenase (HPD) are causally related to two different metabolic disorders: recessively inherited tyrosinemia type III and dominantly inherited hawkinsinuria. The aim of this study was to provide a new perspective for the clinical understanding of the pathogenesis of tyrosinemia type III or hawkinsinuria. Case presentation A full-term newborn baby born after a safe pregnancy and childbirth with a birth weight of 3200 g and another full-term baby born after a safe pregnancy and childbirth with a birth weight of 2800 g are reported and analysed. DNA extraction, next-generation sequencing, bioinformatics analysis, Sanger sequencing and biochemical analysis were performed. One patient with a heterozygous HPD gene (NM_002150.2) c.460G > A mutation and one patient with a heterozygous HPD gene (NM_002150.2) c.248delG mutation showing elevated tyrosine levels upon newborn screening by tandem mass spectrometry (MS/MS) are reported. Conclusions The HPD gene may not be a strictly autosomal recessive pathogenic gene, which provides a new perspective for the clinical understanding of the pathogenesis of tyrosinemia type III or hawkinsinuria.
Hawkinsinuria clinical practice guidelines: a Mexican case report and literature review.
Hawkinsinuria is an autosomal dominant disorder of tyrosine metabolism. Mutations in the 4-hydroxyphenylpyruvate dioxygenase gene (HPD) result in an altered HPD enzyme, causing hawkinsin and tyrosine accumulation. Persistent metabolic acidosis and failure to thrive are common features in patients with hawkinsinuria. We present the first known Latin American patient diagnosed with hawkinsinuria, and the tenth reported patient in the literature. We aim to establish clinical practice guidelines for patients with hawkinsinuria. The patient's plasma tyrosine level was 21.5 mg/dL, which is several times higher than the reference value. Mutation analysis indicated heterozygosity for V212M and A33T variants in HPD. In the case of altered tyrosine levels found during newborn screening, we propose exclusive breastmilk feeding supplemented with ascorbic acid. Amino acid quantification is useful for monitoring treatment response. If tyrosinemia persists, protein intake must be decreased via a low-tyrosine diet. Molecular studies can be used to confirm a patient's disease etiology. Further reports are required to elucidate new pathogenic and phenotypic variations to enable the development of an appropriate therapeutic approach.
Publicações recentes
Challenges in diagnosing hawkinsinuria in adulthood: 2 cases from a single family.
🥉 Relato de casoMolecular and Evolution In Silico Studies Unlock the h4-HPPD C-Terminal Tail Gating Mechanism.
A sportomics soccer investigation unveils an exercise-induced shift in tyrosine metabolism leading to hawkinsinuria.
Variant analysis of HPD genes from two families showing elevated tyrosine upon newborn screening by tandem mass spectrometry (MS/MS).
🥉 Relato de casoHawkinsinuria clinical practice guidelines: a Mexican case report and literature review.
🥉 Relato de caso📚 EuropePMC16 artigos no totalmostrando 10
Challenges in diagnosing hawkinsinuria in adulthood: 2 cases from a single family.
BMJ case reportsMolecular and Evolution In Silico Studies Unlock the h4-HPPD C-Terminal Tail Gating Mechanism.
BiomedicinesA sportomics soccer investigation unveils an exercise-induced shift in tyrosine metabolism leading to hawkinsinuria.
Frontiers in nutritionVariant analysis of HPD genes from two families showing elevated tyrosine upon newborn screening by tandem mass spectrometry (MS/MS).
Journal of pediatric endocrinology & metabolism : JPEMHawkinsinuria clinical practice guidelines: a Mexican case report and literature review.
The Journal of international medical researchIn-Silico analysis of missense SNPs in Human HPPD gene associated with Tyrosinemia type III and Hawkinsinuria.
Computational biology and chemistryHawkinsinuria With Direct Hyperbilirubinemia in Egyptian-Lebanese Boy.
Frontiers in pediatricsExpanding the phenotype of hawkinsinuria: new insights from response to N-acetyl-L-cysteine.
Journal of inherited metabolic diseaseQualitative urinary organic acid analysis: 10 years of quality assurance.
Journal of inherited metabolic diseaseHawkinsinuria in two unrelated Greek newborns: identification of a novel variant, biochemical findings and treatment.
Journal of pediatric endocrinology & metabolism : JPEMAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Hawkinsinúria.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Hawkinsinúria
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Challenges in diagnosing hawkinsinuria in adulthood: 2 cases from a single family.
- Molecular and Evolution In Silico Studies Unlock the h4-HPPD C-Terminal Tail Gating Mechanism.
- A sportomics soccer investigation unveils an exercise-induced shift in tyrosine metabolism leading to hawkinsinuria.
- Variant analysis of HPD genes from two families showing elevated tyrosine upon newborn screening by tandem mass spectrometry (MS/MS).
- Hawkinsinuria clinical practice guidelines: a Mexican case report and literature review.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2118(Orphanet)
- OMIM OMIM:140350(OMIM)
- MONDO:0007700(MONDO)
- GARD:5668(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q5685180(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
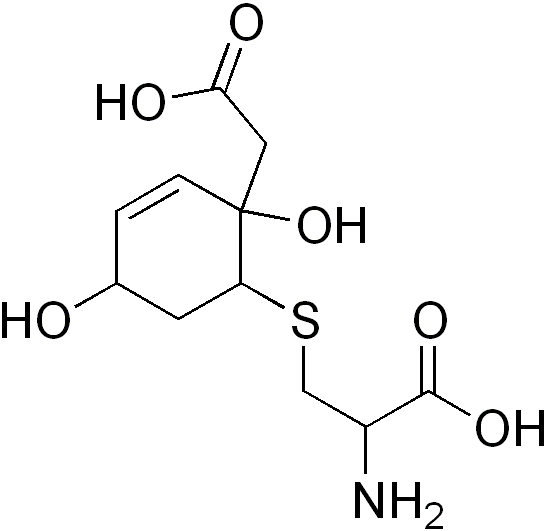
Hawkinsinúria
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata