Síndrome rara causada pela exclusão de material genético no braço curto do cromossomo 17. É caracterizada por um cérebro anormalmente liso com menos dobras e sulcos. Resulta em deficiência intelectual, atraso no desenvolvimento, convulsões, espasticidade, hipotonia e dificuldades alimentares. Os indivíduos afetados têm características faciais distintas que incluem testa proeminente, hipoplasia do terço médio da face, nariz pequeno e arrebitado, orelhas de inserção baixa, mandíbula pequena e lábio superior grosso.
Introdução
O que você precisa saber de cara
Síndrome rara causada pela exclusão de material genético no braço curto do cromossomo 17. É caracterizada por um cérebro anormalmente liso com menos dobras e sulcos. Resulta em deficiência intelectual, atraso no desenvolvimento, convulsões, espasticidade, hipotonia e dificuldades alimentares. Os indivíduos afetados têm características faciais distintas que incluem testa proeminente, hipoplasia do terço médio da face, nariz pequeno e arrebitado, orelhas de inserção baixa, mandíbula pequena e lábio superior grosso.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 24 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 61 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
3 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant.
Adapter protein implicated in the regulation of a large spectrum of both general and specialized signaling pathways (PubMed:21189250). Binds to a large number of partners, usually by recognition of a phosphoserine or phosphothreonine motif (PubMed:35343654). Binding generally results in the modulation of the activity of the binding partner (By similarity). Positively regulates phosphorylated protein HSF1 nuclear export to the cytoplasm (PubMed:12917326). Plays a positive role in the antiviral si
NucleusCytoplasmMelanosome
Transcriptional repressor (PubMed:12052894, PubMed:15231840). Recognizes and binds to the consensus sequence '5-[CG]NG[CG]GGGCA[CA]CC-3' (PubMed:15231840). May act as a tumor suppressor (PubMed:20154726). Involved in development of head, face, limbs and ventral body wall (By similarity). Involved in down-regulation of SIRT1 and thereby is involved in regulation of p53/TP53-dependent apoptotic DNA-damage responses (PubMed:16269335). The specific target gene promoter association seems to be depend
Nucleus
Regulatory subunit (beta subunit) of the cytosolic type I platelet-activating factor (PAF) acetylhydrolase (PAF-AH (I)), an enzyme that catalyzes the hydrolyze of the acetyl group at the sn-2 position of PAF and its analogs and participates in PAF inactivation. Regulates the PAF-AH (I) activity in a catalytic dimer composition-dependent manner (By similarity). Required for proper activation of Rho GTPases and actin polymerization at the leading edge of locomoting cerebellar neurons and postmigra
Cytoplasm, cytoskeletonCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleNucleus membrane
Lissencephaly 1
A classical lissencephaly. It is characterized by agyria or pachygyria and disorganization of the clear neuronal lamination of normal six-layered cortex. The cortex is abnormally thick and poorly organized with 4 primitive layers. Associated with enlarged and dysmorphic ventricles and often hypoplasia of the corpus callosum.
Variantes genéticas (ClinVar)
495 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 3 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
29 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Lisencefalia tipo 1 por anomalias no gene LIS 1
Centros de Referência SUS
24 centros habilitados pelo SUS para Lisencefalia tipo 1 por anomalias no gene LIS 1
Centros para Lisencefalia tipo 1 por anomalias no gene LIS 1
Detalhes dos centros
Hospital Universitário Prof. Edgard Santos (HUPES)
R. Dr. Augusto Viana, s/n - Canela, Salvador - BA, 40110-060 · CNES 0003808
Serviço de Referência
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFG
Rua 235 QD. 68 Lote Área, Nº 285, s/nº - Setor Leste Universitário, Goiânia - GO, 74605-050 · CNES 2338424
Serviço de Referência
Hospital Universitário da UFJF
R. Catulo Breviglieri, Bairro - s/n - Santa Catarina, Juiz de Fora - MG, 36036-110 · CNES 2297442
Atenção Especializada
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário Julio Müller (HUJM)
R. Luis Philippe Pereira Leite, s/n - Alvorada, Cuiabá - MT, 78048-902 · CNES 2726092
Atenção Especializada
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Hospital Universitário Lauro Wanderley (HULW)
R. Tabeliao Estanislau Eloy, 585 - Castelo Branco, João Pessoa - PB, 58050-585 · CNES 0002470
Atenção Especializada
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital Universitário Regional de Maringá (HUM)
Av. Mandacaru, 1590 - Parque das Laranjeiras, Maringá - PR, 87083-240 · CNES 2216108
Atenção Especializada
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Hospital Universitário Pedro Ernesto (HUPE-UERJ)
Blvd. 28 de Setembro, 77 - Vila Isabel, Rio de Janeiro - RJ, 20551-030 · CNES 2280221
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital São Lucas da PUCRS
Av. Ipiranga, 6690 - Jardim Botânico, Porto Alegre - RS, 90610-000 · CNES 2232928
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital Universitário da UFSC (HU-UFSC)
R. Profa. Maria Flora Pausewang - Trindade, Florianópolis - SC, 88036-800 · CNES 2560356
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Base de São José do Rio Preto
Av. Brg. Faria Lima, 5544 - Vila Sao Jose, São José do Rio Preto - SP, 15090-000 · CNES 2079798
Atenção Especializada
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
UNIFESP / Hospital São Paulo
R. Napoleão de Barros, 715 - Vila Clementino, São Paulo - SP, 04024-002 · CNES 2688689
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
0 ensaios clínicos encontrados.
Publicações mais relevantes
Long-term outcome in children with infantile epileptic spasms syndrome: a multicenter retrospective study in Korea.
Infantile epileptic spasms syndrome (IESS) is a severe form of infantile epilepsy with a high lifetime morbidity burden. We aimed to assess the long-term epilepsy and neurodevelopmental outcomes based on how children with IESS have been managed over the past few decades. This retrospective multicenter study included children diagnosed with IESS between 1994 and 2021 with a minimum follow-up period of 2 years. Data on demographics, clinical features, medical history, diagnostic evaluations, and treatments used to control spasms were collected. Epilepsy and neurodevelopmental outcomes were assessed at final follow-up. A total of 378 infants with IESS were included. The mean age at onset of spasms was 7.3 (range, 1-24) months and mean follow-up duration was 7.9 (range, 2-28) years. Etiologies were identified in 65.1% of cases, with acquired structural etiologies being the most prevalent (29.9%). Among the genetic and genetic-structural etiologies, tuberous sclerosis complex (n=35), Down syndrome (n=8), Miller-Dieker syndrome (n=3), and 15q duplication syndrome (n=3) were the most common. Vigabatrin was prescribed to 93.9% of the patients, suggesting that it was the mainstay of treatment. At the last follow-up, 77.8% of the children remained on antiseizure medications and 29.1% had drug-resistant epilepsy. Approximately 90% had intellectual disabilities, and half of the eligible individuals had received special education. The IESS imposes a substantial burden on affected children and their families and often leads to chronic epilepsy and impaired cognitive function. Consensus diagnostic and treatment guidelines tailored to the Korean clinical practice are necessary to ensure early diagnosis and timely treatment.
Understanding the Molecular Basis of Miller-Dieker Syndrome.
Miller-Dieker Syndrome (MDS) is a rare neurodevelopmental disorder caused by a heterozygous deletion of approximately 26 genes within the MDS locus of human chromosome 17. MDS, which affects 1 in 100,000 babies, can lead to a range of phenotypes, including lissencephaly, severe neurological defects, distinctive facial abnormalities, cognitive impairments, seizures, growth retardation, and congenital heart and liver abnormalities. One hallmark feature of MDS is an unusually smooth brain surface due to abnormal neuronal migration during early brain development. Several genes located within the MDS locus have been implicated in the pathogenesis of MDS, including PAFAH1B1, YWHAE, CRK, and METTL16. These genes play a role in the molecular and cellular pathways that are vital for neuronal migration, the proper development of the cerebral cortex, and protein translation in MDS. Improved model systems, such as MDS patient-derived organoids and multi-omics analyses indicate that WNT/β-catenin signaling, calcium signaling, S-adenosyl methionine (SAM) homeostasis, mammalian target of rapamycin (mTOR) signaling, Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling, and others are dysfunctional in MDS. This review of MDS integrates details at the clinical level alongside newly emerging details at the molecular and cellular levels, which may inform the development of novel therapeutic strategies for MDS.
Diagnosis of Lissencephaly in a Neonate After Antenatal Polyhydramnios and Suspicion of Fetal Esophageal Atresia: A Case Report.
Polyhydramnios is a non-specific prenatal finding associated with several fetal anomalies, most notably gastrointestinal obstructions, such as esophageal atresia (EA). However, its etiology may extend beyond gastrointestinal causes to include central nervous system (CNS) abnormalities. We report the case of a neonate initially suspected in utero to have EA based on polyhydramnios and a small fetal gastric bubble, but postnatally diagnosed with classical lissencephaly associated with Miller-Dieker syndrome. A 32-year-old primigravida was referred to our center at 28 weeks of gestation on account of suspected fetal EA with polyhydramnios and a small gastric bubble on prenatal ultrasonography (US). No other anomaly was identified, and she was admitted for management at 30 4/7 weeks of gestation. The patient was delivered at 36 3/7 weeks of gestation via emergency cesarean section due to premature rupture of membranes and arrest of vaginal labor. The neonate showed no sign of EA, and that diagnosis was excluded based on successful passage of an orogastric tube and normal abdominal radiography. Mild dysmorphic features, such as micrognathia and hypertelorism, were noted. As the etiology of polyhydramnios remained unclear, neuroimaging was performed. Cranial US findings were unremarkable; however, head magnetic resonance imaging (MRI) on day seven of life revealed a thickened cortex lacking normal sulcation, which is consistent with classical lissencephaly. Chromosomal microarray analysis revealed a deletion on 17p13.3, confirming Miller-Dieker syndrome. The patient was discharged on day 35 after an uneventful neonatal period; however, epileptic spasms and developmental delay were noticed from the age of six months. This case highlights the diagnostic challenge of differentiating between gastrointestinal and neurological causes of polyhydramnios. Although EA is a common differential diagnosis in such cases, the potential for CNS malformations mimicking gastrointestinal pathologies must be considered. Routine fetal ultrasound alone may fail to detect subtle cerebral abnormalities such as lissencephaly, especially in the absence of ventriculomegaly or other signs. Fetal MRI, which has superior soft tissue resolution, should be considered when the etiology of polyhydramnios is uncertain, even if there is no overt CNS anomaly. This case highlights the importance of prompt neuroimaging, including fetal MRI, which may facilitate diagnosis and counseling of families, and optimize postnatal care.
Deep clinical and genetic analysis of 17p13.3 region: 38 pediatric patients diagnosed using next-generation sequencing and literature review.
Chromosome 17p13.3 is a region of genomic instability associated with different neurodevelopmental diseases. The malformation spectrum of 17p13.3 microdeletions ranges from an isolated lissencephaly sequence to Miller-Dieker syndrome, while 17p13.3 microduplications result in autism, learning disabilities, microcephaly and other brain malformations. This study aims to provide a more comprehensive delineation of the clinical and genetic characteristics associated with 17p13.3 alterations. We retrospectively analyzed the next-generation sequencing (NGS) data of more than 40 thousand patients from January 2016 to December 2021 and identified 38 pediatric patients with copy-number variations (CNVs) or single-nucleotide variations (SNVs) in 17p13.3 region. Published patients with CNVs in the 17p13.3 region were also collected and we performed a Chi-square test to compare the phenotype spectrum of microdeletions and microduplications. Among the 27 CNV patients, 20 patients with microdeletions and 7 patients with microduplications were found. PAFAH1B1 was the most frequently deleted gene and CRK was the most frequently duplicated gene. Affected genes in 11 SNV patients included PAFAH1B1 and PRPF8. Developmental delay was the most common abnormality detected in the 38 patients (29/38, 76.3%). Of note, Case 10 presented omphalocele and Case 23 presented scoliosis, webbed neck and bone cyst, all of which were unusual variant phenotypes in this region. The Chi-square test revealed that epilepsy, lissencephaly and short stature were statistically significant with microdeletions, while behavioral abnormalities and hand and foot abnormalities were significant with microduplications (p < 0.01). While PAFAH1B1, YWHAE and CRK are associated with major phenotypes of 17p13.3, RTN4RL1 may be involved in white matter changes and HIC1 might contribute to the occurrence of omphalocele. This study provided a comprehensive understanding of genetic information and phenotype spectrum of the 17p13.3 region.
Ring Chromosome 17 Syndrome-A Case Report and Discussion of Diagnostic Methods.
Ring chromosome 17 and 17p13.3 deletion syndrome are phenotypically heterogeneous diseases with similar clinical features. The ring chromosome 17 phenotypic features range from the Miller-Dieker syndrome characterized by deletion of the PAFAH1B1 gene, lissencephaly, hypotonia, dysphagia, café au lait spots, and severe intellectual disability, to a milder phenotype characterized by microcephaly, seizures, delayed development, minor facial dysmorphic features, clinodactyly, short stature, café au lait spots, retinal flecking, and deletion of the YWHAE and CRK genes. Similarly, the phenotypic features of the 17p13.3 deletion syndrome range from the Miller-Dieker syndrome caused by loss of function of the PAFAH1B1 gene and characterized by lissencephaly, microcephaly, seizures, hypotonia, and severe intellectual disability to a milder phenotype characterized by nonspecific white matter changes, microcephaly, seizures, delayed development, short stature, and deletion of the YWHAE and CRK genes. Café au lait spots and retinal or axillary freckling have been noted in the ring chromosome 17 syndrome but not in 17p13.3 deletion syndrome. We report a 5-year-old girl with a history of intrauterine growth retardation, short stature, intractable epilepsy, expressive language disorder, clinodactyly, multiple café au lait spots, and retinal freckling who was initially diagnosed with 17p13.3 deletion syndrome involving YWHAE and CRK but not PAFAH1B1 on CGH array. However, cytogenetic analysis of G-banded chromosomes revealed mosaic ring chromosome 17. Optical genome mapping simultaneously identified the 17p13.3 deletion and the mosaic ring chromosome 17. This case report highlights the limitations of the arrays and sequencing methods for identifying structural variants, the need to investigate further deletions and duplications identified by arrays, mainly considering atypical phenotypic features, and suggests that OGM could be used as a first-tier test with exome sequencing for the diagnosis of patients with dysmorphic features, intellectual disability, and seizure disorder.
Publicações recentes
Miller-Dieker Syndrome: Genetic Etiology, Neurocognitive Impact, and Clinical Implications in a Neuronal Migration Disorder.
Long-term outcome in children with infantile epileptic spasms syndrome: a multicenter retrospective study in Korea.
Diagnosis of Lissencephaly in a Neonate After Antenatal Polyhydramnios and Suspicion of Fetal Esophageal Atresia: A Case Report.
Understanding the Molecular Basis of Miller-Dieker Syndrome.
Deep clinical and genetic analysis of 17p13.3 region: 38 pediatric patients diagnosed using next-generation sequencing and literature review.
📚 EuropePMC98 artigos no totalmostrando 58
Long-term outcome in children with infantile epileptic spasms syndrome: a multicenter retrospective study in Korea.
Clinical and experimental pediatricsDiagnosis of Lissencephaly in a Neonate After Antenatal Polyhydramnios and Suspicion of Fetal Esophageal Atresia: A Case Report.
CureusUnderstanding the Molecular Basis of Miller-Dieker Syndrome.
International journal of molecular sciencesDeep clinical and genetic analysis of 17p13.3 region: 38 pediatric patients diagnosed using next-generation sequencing and literature review.
BMC medical genomicsRing Chromosome 17 Syndrome-A Case Report and Discussion of Diagnostic Methods.
American journal of medical genetics. Part AMulti-Omics Approach Reveals Genes and Pathways Affected in Miller-Dieker Syndrome.
Molecular neurobiologyTotal callosotomy ameliorates epileptic activity and improves cognitive function in a patient with Miller-Dieker syndrome.
Epilepsy & behavior reportsRETRACTED: Bahmad et al. Histopathologic Findings Associated with Miller-Dieker Syndrome: An Autopsy Report. Diseases 2022, 10, 95.
Diseases (Basel, Switzerland)Acute Bowel Ischemia in a Premature Neonate with Miller-Dieker Syndrome and Anomalous Right Coronary Artery From the Pulmonary Artery.
Pediatric annalsCrk/Crkl regulates early angiogenesis in mouse embryos by accelerating endothelial cell maturation.
bioRxiv : the preprint server for biologyAnesthetic Management and Bispectral Index in a Child with Miller-Dieker Syndrome: A Case Report.
Children (Basel, Switzerland)YWHAE loss of function causes a rare neurodevelopmental disease with brain abnormalities in human and mouse.
Genetics in medicine : official journal of the American College of Medical Genetics[Prenatal genetic analysis of a fetus with Miller-Dieker syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsClinical findings and genetic analysis of patients with copy number variants involving 17p13.3 using a single nucleotide polymorphism array: a single-center experience.
BMC medical genomicsFurther expansion and confirmation of phenotype in rare loss of YWHAE gene distinct from Miller-Dieker syndrome.
American journal of medical genetics. Part AHistopathologic Findings Associated with Miller-Dieker Syndrome: An Autopsy Report.
Diseases (Basel, Switzerland)Genetic testing in adults with developmental and epileptic encephalopathy - what do we know?
Medizinische Genetik : Mitteilungsblatt des Berufsverbandes Medizinische Genetik e.VCentral-part laryngectomy after laryngotracheal separation to manage pharyngocutaneous fistula: A case report and retrospective analysis of 12 cases.
Auris, nasus, larynxPrenatal diagnosis of Miller-Dieker syndrome/PAFAH1B1-related lissencephaly: Ultrasonography and genetically investigative results.
European journal of obstetrics, gynecology, and reproductive biologyApplication of Interphase Fluorescent in Situ Hybridization: a Screening Tool for the Diagnosis of Microdeletion Syndrome.
Clinical laboratoryUnusual presentation of acute encephalopathy with biphasic seizures and late reduced diffusion in Miller-Dieker syndrome.
BMJ case reportsCrk Haploinsufficiency Is Associated with Intrauterine Growth Retardation and Severe Postnatal Growth Failure.
Hormone research in paediatricsResponsible Genes for Neuronal Migration in the Chromosome 17p13.3: Beyond Pafah1b1(Lis1), Crk and Ywhae(14-3-3ε).
Brain sciencesRpsa Signaling Regulates Cortical Neuronal Morphogenesis via Its Ligand, PEDF, and Plasma Membrane Interaction Partner, Itga6.
Cerebral cortex (New York, N.Y. : 1991)BACs-on-Beads Assay for the Prenatal Diagnosis of Microdeletion and Microduplication Syndromes.
Molecular diagnosis & therapy[Prenatal diagnosis and genetic analysis of a fetus with Miller-Dieker syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPatient-derived iPSC modeling of rare neurodevelopmental disorders: Molecular pathophysiology and prospective therapies.
Neuroscience and biobehavioral reviews[Prenatal diagnosis of a fetus with Miller-Dieker syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsPrenatal diagnosis of Miller-Dieker syndrome by chromosomal microarray.
Annals of human geneticsPrenatal diagnosis of BACs-on-Beads assay in 1520 cases from Fujian Province, China.
Molecular genetics & genomic medicineIn vitro modeling for inherited neurological diseases using induced pluripotent stem cells: from 2D to organoid.
Archives of pharmacal researchThe transcription factor Hypermethylated in Cancer 1 (Hic1) regulates neural crest migration via interaction with Wnt signaling.
Developmental biology17p13.3 microdeletion including YWHAE and CRK genes: towards a clinical characterization.
Neurological sciences : official journal of the Italian Neurological Society and of the Italian Society of Clinical NeurophysiologyNeuronal migration genes and a familial translocation t (3;17): candidate genes implicated in the phenotype.
BMC medical geneticsRare Concurrence of Two Congenital Disorders: Miller-Dieker Syndrome and T-Cell Lymphopenia.
Cytogenetic and genome researchPerampanel in lissencephaly-associated epilepsy.
Epilepsy & behavior case reportsMicrodeletions excluding YWHAE and PAFAH1B1 cause a unique leukoencephalopathy: further delineation of the 17p13.3 microdeletion spectrum.
Genetics in medicine : official journal of the American College of Medical GeneticsPrenatal Diagnosis of BACs-on-Beads Assay in 3647 Cases of Amniotic Fluid Cells.
Reproductive sciences (Thousand Oaks, Calif.)Electroclinical Pattern and Epilepsy Evolution in an Infant with Miller-Dieker Syndrome.
Journal of pediatric neurosciences17p13.3 quadruplication: a prenatal and postpartum clinical characterization of a copy number variant.
Cold Spring Harbor molecular case studiesCase Report of Proliferative Peripheral Retinopathy in Two Familial Lissencephaly Infants with Miller-Dieker Syndrome.
Journal of pediatric geneticsNeurodevelopmental Genetic Diseases Associated With Microdeletions and Microduplications of Chromosome 17p13.3.
Frontiers in geneticsApplication of the BACs-on-Beads™ assay for rapid prenatal detection application of BoBs™ for PND of aneuploidies and microdeletions.
Molecular reproduction and development[Application of chromosomal microarray analysis for fetuses with ventricular septal defects].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsMiller-Dieker Syndrome due to a 5.5-Mb 17p Deletion in a 17;Y Pseudodicentric Chromosome.
Cytogenetic and genome researchDisruption of YWHAE gene at 17p13.3 causes learning disabilities and brain abnormalities.
Clinical geneticsAn Organoid-Based Model of Cortical Development Identifies Non-Cell-Autonomous Defects in Wnt Signaling Contributing to Miller-Dieker Syndrome.
Cell reports[Prenatal genetic analysis of two fetuses with Miller-Dieker syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsHuman iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia.
Cell stem cellMiller-Dieker Syndrome with unbalanced translocation 45, X, psu dic(17;Y)(p13;p11.32) detected by fluorescence in situ hybridization and G-banding analysis using high resolution banding technique.
Congenital anomaliesDe novo large rare copy-number variations contribute to conotruncal heart disease in Chinese patients.
NPJ genomic medicineA Case of Concurrent Miller-Dieker Syndrome (17p13.3 Deletion) and 22q11.2 Deletion Syndrome.
Journal of pediatric genetics[Prenatal diagnosis of chromosome abnormalities and nine microdeletion syndromes using both traditional karyotyping and BoBs].
Zhonghua fu chan ke za zhi[17p13.3 duplication as a cause of psychomotor developmental delay in an infant - a further case of a new syndrome].
Polski merkuriusz lekarski : organ Polskiego Towarzystwa LekarskiegoCharacterization of intragenic tandem duplication in the PAFAH1B1 gene leading to isolated lissencephaly sequence.
Molecular cytogeneticsRoutine chromosomal microarray analysis is necessary in Korean patients with unexplained developmental delay/mental retardation/autism spectrum disorder.
Annals of laboratory medicineProtein-Protein and Peptide-Protein Interactions of NudE-Like 1 (Ndel1): A Protein Involved in Schizophrenia.
Current protein & peptide scienceManagement of general anesthesia in a child with Miller-Dieker syndrome: a case report.
JA clinical reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Lisencefalia tipo 1 por anomalias no gene LIS 1.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Lisencefalia tipo 1 por anomalias no gene LIS 1
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Long-term outcome in children with infantile epileptic spasms syndrome: a multicenter retrospective study in Korea.
- Understanding the Molecular Basis of Miller-Dieker Syndrome.
- Diagnosis of Lissencephaly in a Neonate After Antenatal Polyhydramnios and Suspicion of Fetal Esophageal Atresia: A Case Report.
- Deep clinical and genetic analysis of 17p13.3 region: 38 pediatric patients diagnosed using next-generation sequencing and literature review.
- Ring Chromosome 17 Syndrome-A Case Report and Discussion of Diagnostic Methods.
- Miller-Dieker Syndrome: Genetic Etiology, Neurocognitive Impact, and Clinical Implications in a Neuronal Migration Disorder.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:531(Orphanet)
- OMIM OMIM:247200(OMIM)
- MONDO:0009532(MONDO)
- GARD:3669(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q2200977(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
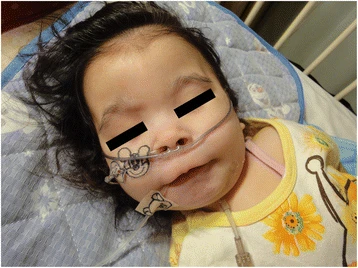
Lisencefalia tipo 1 por anomalias no gene LIS 1
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata