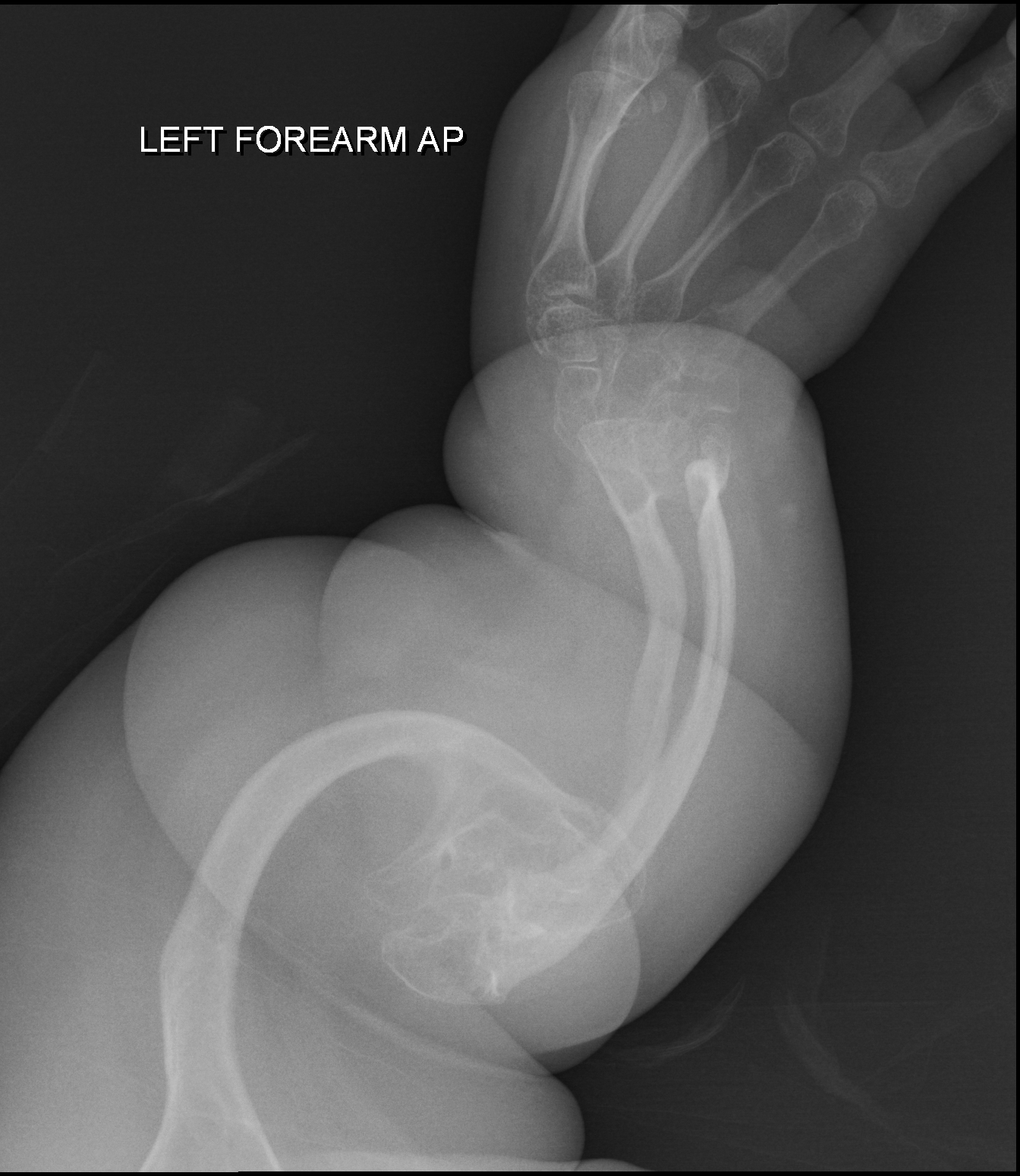
As síndromes de costela curta e polidactilia são um grupo de malformações ósseas (problemas na formação dos ossos). Elas são caracterizadas por um tórax (peito) estreito e por polidactilia, que é a presença de dedos extras, geralmente no lado do polegar ou do dedão do pé.
Introdução
O que você precisa saber de cara
As síndromes de costela curta e polidactilia são um grupo de malformações ósseas (problemas na formação dos ossos). Elas são caracterizadas por um tórax (peito) estreito e por polidactilia, que é a presença de dedos extras, geralmente no lado do polegar ou do dedão do pé.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 205 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 524 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
27 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
May play a role in cell-cycle-dependent microtubule organization
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, spindle poleCell projection, cilium
Joubert syndrome 21
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Phosphorylates a large number of substrates in the cytoplasm and the nucleus (PubMed:15642694, PubMed:15905176, PubMed:16387847, PubMed:17333334, PubMed:17565987, PubMed:17693412, PubMed:18836454, PubMed:19949837, PubMed:20356841, PubMed:21085490, PubMed:21514275, PubMed:21812984, PubMed:21852232, PubMed:31112131). Phosphorylates CDC25B, ABL1, NFKB1, CLDN3, histone H1.4 (H1-4), PSMC5/RPT6, PJA2, RYR2, RORA, SOX9, UHRF1 and VASP (PubMed:15178447, PubMed:15642694, PubMed:15905176, PubMed:16387847,
CytoplasmCell membraneMembraneNucleusMitochondrionCell projection, cilium, flagellumCytoplasmic vesicle, secretory vesicle, acrosome
Primary pigmented nodular adrenocortical disease 4
A rare bilateral adrenal defect causing ACTH-independent Cushing syndrome. Macroscopic appearance of the adrenals is characteristic with small pigmented micronodules observed in the cortex. Adrenal glands show overall normal size and weight, and multiple small yellow-to-dark brown nodules surrounded by a cortex with a uniform appearance. Microscopically, there are moderate diffuse cortical hyperplasia with mostly nonpigmented nodules, multiple capsular deficits and massive circumscribed and infiltrating extra-adrenal cortical excrescences with micronodules. Clinical manifestations of Cushing syndrome include facial and truncal obesity, abdominal striae, muscular weakness, osteoporosis, arterial hypertension, diabetes.
Mediates cAMP-dependent signaling triggered by receptor binding to GPCRs (PubMed:12420224, PubMed:21423175, PubMed:31112131). PKA activation regulates diverse cellular processes such as cell proliferation, the cell cycle, differentiation and regulation of microtubule dynamics, chromatin condensation and decondensation, nuclear envelope disassembly and reassembly, as well as regulation of intracellular transport mechanisms and ion flux (PubMed:12420224, PubMed:21423175). Regulates the abundance o
CytoplasmCell membraneMembraneNucleus
Cardioacrofacial dysplasia 2
An autosomal dominant disease characterized by dysmorphic facial features, congenital cardiac defects, primarily common atrium or atrioventricular septal defect, and limb anomalies, including short limbs, brachydactyly and postaxial polydactyly. CAFD2 patients may show developmental delay of variable severity, intellectual disability, autistic features and focal seizures.
Plays an inhibitory role on IL13 signaling by binding to IL13RA1. Involved in suppression of IL13-induced STAT6 phosphorylation, transcriptional activity and DNA-binding. Recruits TRAF3 and DISC1 to the microtubules. Involved in kidney development and epithelial morphogenesis. Involved in the regulation of microtubule cytoskeleton organization. Is a negative regulator of microtubule stability, acting through the control of MAP4 levels (PubMed:26487268). Involved in ciliogenesis (By similarity)
Cytoplasm, cytoskeletonCell projection, ciliumCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, cilium basal body
Senior-Loken syndrome 9
A renal-retinal disorder characterized by progressive wasting of the filtering unit of the kidney (nephronophthisis), with or without medullary cystic renal disease, and progressive eye disease. Typically this disorder becomes apparent during the first year of life.
Acts as a transcriptional activator (PubMed:10806483, PubMed:19706761, PubMed:19878745, PubMed:24076122, PubMed:24217340, PubMed:24311597). Binds to the DNA consensus sequence 5'-GACCACCCA-3' (PubMed:2105456, PubMed:24217340, PubMed:8378770). Regulates the transcription of specific genes during normal development (PubMed:19706761). Plays a role in craniofacial development and digital development, as well as development of the central nervous system and gastrointestinal tract. Mediates SHH signal
CytoplasmNucleus
Polydactyly, postaxial, A8
A form of postaxial polydactyly, a condition characterized by the occurrence of supernumerary digits in the upper and/or lower extremities. In postaxial polydactyly type A, the extra digit is well-formed and articulates with the fifth or a sixth metacarpal/metatarsal. PAPA8 is an autosomal recessive condition characterized by the presence of postaxial extra digits (hexadactyly) on the hands and/or the feet.
Phosphorylates serines and threonines, but also appears to possess tyrosine kinase activity (PubMed:20230784). Involved in DNA damage checkpoint control and for proper DNA damage repair (PubMed:20230784). In response to injury that includes DNA damage, NEK1 phosphorylates VDAC1 to limit mitochondrial cell death (PubMed:20230784). May be implicated in the control of meiosis (By similarity). Involved in cilium assembly (PubMed:21211617)
NucleusCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm
Short-rib thoracic dysplasia 6 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Involved in centriole duplication (PubMed:24613305, PubMed:26297806). Positively regulates CEP63 centrosomal localization (PubMed:24613305, PubMed:26297806). Required for WDR62 centrosomal localization and promotes the centrosomal localization of CDK2 (PubMed:24613305, PubMed:26297806). May play a role in cilium assembly
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satelliteCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Orofaciodigital syndrome 15
A form of orofaciodigital syndrome, a group of heterogeneous disorders characterized by malformations of the oral cavity, face and digits, and associated phenotypic abnormalities that lead to the delineation of various subtypes. OFD15 features include facial dysmorphism, lobulated tongue, clefting of the alveolar ridges, left hand postaxial polydactyly, broad right hallux and left hallux duplication, and intermittent respiratory difficulty. Brain anomalies include vermis hypoplasia with molar tooth sign, agenesis of corpus callosum, and ventricular dilation. OFD15 inheritance is autosomal recessive.
Plays a role in the microtubule-dependent coupling of the nucleus and the centrosome. Involved in the processes that regulate centrosome-mediated interkinetic nuclear migration (INM) of neural progenitors and for proper positioning of neurons during brain development. Also implicated in the migration and selfrenewal of neural progenitors. Required for centriole duplication and maturation during mitosis and subsequent ciliogenesis (By similarity). Required for the recruitment of CEP295 to the pro
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 13 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Required for the maintenance and formation of cilia. Plays an indirect role in hedgehog (Hh) signaling, cilia being required for all activity of the hedgehog pathway (By similarity)
Cell projection, cilium
Short-rib thoracic dysplasia 10 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
As a component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in ciliogenesis and ciliary protein trafficking (PubMed:21473986, PubMed:28400947, PubMed:29220510). May promote CASP3 activation and TNF-stimulated apoptosis
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, cilium basal body
Cranioectodermal dysplasia 2
A disorder characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include short stature, dolichocephaly, craniosynostosis, narrow thorax with pectus excavatum, short limbs, brachydactyly, joint laxity, narrow palpebral fissures, telecanthus with hypertelorism, low-set simple ears, everted lower lip, and short neck. Teeth abnormalities include widely spaced, hypoplastic and fused teeth.
As a component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is required in ciliogenesis and ciliary protein trafficking (PubMed:27932497, PubMed:29220510). Involved in cilia formation during neuronal patterning. Acts as a negative regulator of Shh signaling. Required to recruit TULP3 to primary cilia (By similarity)
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Cranioectodermal dysplasia 1
A disorder characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include dolichocephaly (with or without sagittal suture synostosis), scaphocephaly, short stature, limb shortening, short ribs, narrow chest, brachydactyly, renal failure and hepatic fibrosis, small and abnormally shaped teeth, sparse hair, skin laxity and abnormal nails.
Plays a key role in ciliogenesis and embryonic development. Regulator of cilia formation by controlling the organization of the apical actin cytoskeleton and the positioning of the basal bodies at the apical cell surface, which in turn is essential for the normal orientation of elongating ciliary microtubules. Plays a key role in definition of cell polarity via its role in ciliogenesis but not via conversion extension. Has an indirect effect on hedgehog signaling (By similarity). Proposed to fun
CytoplasmCell surfaceCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Short-rib thoracic dysplasia 20 with polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
As a component of IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in ciliogenesis (PubMed:28400947, PubMed:28973684). Involved in retrograde ciliary transport along microtubules from the ciliary tip to the base (PubMed:21378380)
Cytoplasm, cytoskeletonCell projection, cilium
Cranioectodermal dysplasia 3
A disorder primarily characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include craniosynostosis, narrow rib cage, short limbs, brachydactyly, hypoplastic and widely spaced teeth, sparse hair, skin laxity and abnormal nails. Nephronophthisis leading to progressive renal failure, hepatic fibrosis, heart defects, and retinitis pigmentosa have also been described.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system, facilitating the assembly of these organelles (PubMed:29742051). Involved in the regulation of ciliary length (PubMed:26077881, PubMed:26130459)
Golgi apparatusCytoplasmCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 15 with polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs). Essential for retrograde trafficking of IFT-1, IFT-B and GPCRs (PubMed:27932497). Negatively modulates the SHH signal transduction (By similarity)
Cytoplasm, cytoskeleton, cilium axoneme
Involved in ciliogenesis as part of a complex involved in intraflagellar transport (IFT), the bi-directional movement of particles required for the assembly, maintenance and functioning of primary cilia (PubMed:27466190). Required for the anterograde transport of IFT88 (PubMed:27466190)
Cell projection, cilium
Short-rib thoracic dysplasia 16 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Required for ciliogenesis and sonic hedgehog/SHH signaling. Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1. May play a role in early ciliogenesis in the disappearance of centriolar satellites that preceeds ciliary vesicle formation (PubMed:24421332). Involved in regulation of cell intracellular organization. Involved in regulation of cell polarity (By similarity). Required for asymmetrical localization of CEP
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomePhotoreceptor inner segmentCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 23
A mild form of Joubert syndrome, a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Component of the intraflagellar transport (IFT) complex B: together with IFT74, forms a tubulin-binding module that specifically mediates transport of tubulin within the cilium. Binds tubulin via its CH (calponin-homology)-like region (PubMed:23990561). Required for ciliogenesis (PubMed:23990561, PubMed:27666822). Required for proper regulation of SHH signaling (PubMed:27666822). Plays an important role during spermatogenesis by modulating the assembly and elongation of the sperm flagella (By si
Cell projection, ciliumCytoplasmCytoplasm, cytoskeleton, cilium basal body
Short-rib thoracic dysplasia 19 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system. Required for proper retrograde ciliary transport
Dynein axonemal particle
Short-rib thoracic dysplasia 17 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs) (PubMed:20889716, PubMed:22503633). Plays a pivotal role in proper development and function of ciliated cells through its role in ciliogenesis and/or cilium maintenance (PubMed:22503633). Required for the development and maintenance of the outer segments of rod and cone photoreceptor cells. Plays a role in maintenance and the delivery of opsin to
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, cilium
Short-rib thoracic dysplasia 9 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome. SRTD9 is characterized by phalangeal cone-shaped epiphyses, chronic renal disease, nearly constant retinal dystrophy, and mild radiographic abnormality of the proximal femur. Occasional features include short stature, cerebellar ataxia, and hepatic fibrosis.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Involved in endochondral growth and skeletal development
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membrane
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
Component of the intraflagellar transport (IFT) complex B, which is essential for the development and maintenance of motile and sensory cilia
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axoneme
Short-rib thoracic dysplasia 2 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system (PubMed:25205765, PubMed:29742051). DYNC2I2 plays a major role in retrograde ciliary protein trafficking and in ciliogenesis (PubMed:29742051, PubMed:30320547, PubMed:30649997). Required also to maintain a functional transit
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, ciliumCell projection, filopodium
Short-rib thoracic dysplasia 11 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Component of the EvC complex that positively regulates ciliary Hedgehog (Hh) signaling. Plays a critical role in bone formation and skeletal development. May be involved in early embryonic morphogenesis
Cell membraneCytoplasm, cytoskeleton, cilium basal bodyCell projection, ciliumCell projection, cilium membraneNucleus
Ellis-van Creveld syndrome
An autosomal recessive condition characterized by the clinical tetrad of chondrodystrophy, polydactyly, ectodermal dysplasia and cardiac anomalies. Patients manifest short-limb dwarfism, short ribs, postaxial polydactyly, and dysplastic nails and teeth. Congenital heart defects, most commonly an atrioventricular septal defect, are observed in 60% of affected individuals.
As component of the IFT complex A (IFT-A), a complex required for retrograde ciliary transport and entry into cilia of G protein-coupled receptors (GPCRs), it is involved in cilia function and/or assembly (PubMed:20889716). Essential for functional IFT-A assembly and ciliary entry of GPCRs (PubMed:20889716). Associates with the BBSome complex to mediate ciliary transport (By similarity)
Cell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCell projection, cilium, photoreceptor outer segmentCell projection, cilium, flagellum
Cranioectodermal dysplasia 4
A disorder primarily characterized by craniofacial, skeletal and ectodermal abnormalities. Clinical features include craniosynostosis, narrow rib cage, short limbs, brachydactyly, hypoplastic and widely spaced teeth, sparse hair, skin laxity and abnormal nails. Nephronophthisis leading to progressive renal failure, hepatic fibrosis, heart defects, and retinitis pigmentosa have also been described.
Acts as one of several non-catalytic accessory components of the cytoplasmic dynein 2 complex (dynein-2 complex), a motor protein complex that drives the movement of cargos along microtubules within cilia and flagella in concert with the intraflagellar transport (IFT) system (PubMed:23910462, PubMed:25205765, PubMed:29742051, PubMed:31451806). DYNC2I1 plays a major role in retrograde ciliary protein trafficking in cilia and flagella (PubMed:29742051, PubMed:30320547, PubMed:30649997). Also requi
Cell projection, ciliumCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 8 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
May function as a motor for intraflagellar retrograde transport. Functions in cilia biogenesis. May play a role in transport between endoplasmic reticulum and Golgi or organization of the Golgi in cells (By similarity)
Cytoplasm, cytoskeleton, cilium axonemeCell membraneCytoplasm
Short-rib thoracic dysplasia 3 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Variantes genéticas (ClinVar)
280 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 26 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
41 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de costela curta-polidactilia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
A homozygous frameshift variant in the CILK1 gene causes cranioectodermal dysplasia.
Cranioectodermal dysplasia (CED) is a ciliopathy characterized by skeletal and ectodermal abnormalities, renal failure, and liver fibrosis. Pathogenic variants in genes that encode the intraflagellar transport (IFT) complex components, particularly IFT-A, are responsible for approximately two-thirds of the CED cases. However, the cause of the remaining cases remains unknown. Ciliogenesis-associated kinase 1 (CILK1) is a highly conserved ciliary serine/threonine kinase with an N-terminal catalytic domain responsible for kinase activity and a C-terminal non-catalytic domain that interacts with the IFT-B complex. Biallelic variants in the catalytic domain are associated with lethal skeletal dysplasia, endocrine cerebroosteodysplasia, and short-rib polydactyly syndrome. No human disease has been linked to biallelic variants in the non-catalytic domain. We present a homozygous frameshift variant in the CILK1 gene that affects the distal part of the non-catalytic domain, causing CED in five patients from two pedigrees. All the patients survived into childhood and had disproportionately short stature, skeletal abnormalities, ectodermal dysplasia, renal issues, and liver complications. Functional data from patient-derived cells and the C. elegans model indicate that the variant reduces cilia number, increases cilia length, and disrupts the localization of IFT components. In contrast, the ciliary localization of CILK1 bearing the variant itself remains unaffected. Notably, we rescued the majority of these abnormalities by reintroducing CILK1 into patient-derived cells. Finally, our study describes CILK1 as a novel causal gene and the first non-IFT protein-encoding gene in the etiology of CED, thus expanding the known genotypic, mechanistic, and phenotypic spectrum of CED.
Complete loss of IFT27 function leads to a phenotypic spectrum of fetal lethal ciliopathy associated with altered ciliogenesis.
Ciliopathies are rare genetic diseases marked by considerable phenotypic heterogeneity and overlap. Among the key mechanisms of cilium biology, its compartmentalization is achieved through gating complexes and active transport such as intraflagellar transport (IFT). Among the IFT components, IFT27 plays a role in BBSome-mediated transport of ciliary membrane proteins required for ciliary signaling. While this gene was first linked to Bardet-Biedl syndrome, we next expanded its phenotypic spectrum to a fetal lethal ciliopathy. Here, we identified a second fetal case with short ribs, polydactyly, hypodysplastic kidneys, imperforate anus, and situs inversus. Genome sequencing identified novel biallelic variants in IFT27. Functional analysis of tissues from both fetal cases revealed that all the identified variants lead to mRNA decay. Immunohistochemistry on fetal kidney sections showed that those variants are associated with altered ciliogenesis. Overall, we showed that complete loss of IFT27 function leads to a severe phenotypic spectrum overlapping with short ribs polydactyly and Pallister-Hall syndromes. In addition, our results argue for a role of IFT27 in ciliogenesis in humans.
[Clinical characteristics and prenatal diagnosis of a fetus with Short-rib thoracic dysplasia syndrome due to variants of DYNC2H1 gene].
To explore the prenatal features and genetic etiology of a fetus with Short-rib cage dysplasia (SRTD) due to variants of DYNC2H1 gene. A pregnant women presented at Xinxiang Central Hospital in June 2020 for abnormal prenatal ultrasound findings was selected as the study subject. With informed consent obtained, amniotic fluid sample was extracted from the woman, and clinical data of the fetus were collected. Whole exome sequencing (WES) was carried out, and candidate variants were verified by Sanger sequencing. This study was approved by the Medical Ethics Committee of Xinxiang Central Hospital [Ethics No.: 2025-214-01(K)]. At 25+6 weeks gestation, genetic testing revealed that the fetus has harbored compound heterozygous variants of the DYNC2H1 gene, namely c.10585C>T (p.Arg3529Ter) and c.8954T>G (p.Val2985Gly), which were derived from its father and mother, respectively. Based on the guidelines from the American College of Medical Genetics and Genomics (ACMG), the c.10585C>T (p.Arg3529Ter) and c.8954T>G (p.Val2985Gly) variants were classified as pathogenic (PVS1+PM2_supporting+PM3+PP5) and likely pathogenic (PM1+PM2_supporting+PM3+PP3), respectively. Bioinformatics analysis suggested that both variants may affect the 3D structure of the DYNC2H1 protein. The compound heterozygous variants of c.10585C>T (p.Arg3529Ter) and c.8954T>G (p.Val2985Gly) of the DYNC2H1 gene probably underlay the pathogenesis of SRTD in the fetus. Above findings had facilitated prenatal diagnosis and genetic counseling for the couple.
[Clinical analysis of a patient of Short rib-polydactyly syndrome type 6 with long term misdiagnosis].
To analyze the clinical characteristics of a patient with Short rib-polydactyly syndrome type 6 (SRTD6) with long-term misdiagnosis, and improve its clinical recognition by reviewing the relevant literature. A patient presented at the Children's Hospital Affiliated to Zhejiang University School of Medicine on August 19, 2024 for the discovery of liver dysfunction for 13 years and vision loss for 9 years was selected as the study subject. Her medical history, clinical data, laboratory findings and results of imaging examination were collected. High-throughput sequencing was carried out, and candidate variants were verified by Sanger sequencing. This study was approved by the Ethics Committee of the Hospital (Ethics No.: 2021-IRB-292). The patient had long-term unexplained liver dysfunction, vision loss, and growth delay. Blood acylcarnitine and urinary organic acid analysis have failed to found any abnormality. Previous genetic testing revealed a homozygous c.203A>C (p.Glu68Ala) missense variant in the ETFDH gene, leading to a misdiagnosis of various acyl-CoA dehydrogenase deficiencies. However, treatment with high-dose vitamin B2 showed a poor effect. Physical examination revealed small hands, short and stubby fingers, and a narrow chest. Medical imaging showed shortened bilateral ribs, a narrowed chest, and short, thick metacarpals. High-throughput sequencing has detected a pathogenic homozygous c.1957C>T (p.R653*) nonsense variant in the NEK1 gene, confirming the diagnosis of SRTD6. SRTD6 is characterized by rib and sternum dysplasia as the primary skeletal deformities, which is often accompanied by multi-organ impairment. Genetic testing can facilitate the precise diagnosis.
[Two cases of skeletal ciliopathies in one family].
Cilia are thin extensions on the cells of eukaryotic organisms. They are formed by a special protein transport mechanism - the intraflagellar transporter (IFT). The IFT consists of two proteins: complex A and complex B. Mutations in the genes of the IFT-A complex (IFT43, IFT121, IFT122, IFT139, IFT140, and IFT144) lead to the development of skeletal ciliopathies. These include Sensenbrenner, Jeune, and short-rib polydactyly syndrome [1,2]. We report two cases of different ciliopathies in a non-related family; both parents are heterozygous carriers of a pathogenic mutation in the IFT122 gene. Als Zilien werden dünne Fortsätze auf den Zellen von eukaryoten Organismen bezeichnet. Diese werden von einem speziellen Proteintransportmechanismus – dem intraflagellar Transporter (IFT) gebildet. Der IFT besteht aus zwei Proteinen: Komplex A und Komplex B. Mutationen in den Genen des IFT-A-Komplexes (IFT43, IFT121, IFT122, IFT139, IFT140 und IFT144) führen zur Entstehung von Skelett-Ziliopathien. Zu diesen zählen das Sensenbrenner-, Jeune- und Kurzrippen-Polydaktylie-Syndrom [1,2]. Wir berichten über zwei Fälle unterschiedlicher Ziliopathien in einer nicht-blutsverwandten Familie, beide Elternteile sind heterozygote Träger einer pathogenen Mutation im IFT122- Gen.
Publicações recentes
[Clinical analysis of a patient of Short rib-polydactyly syndrome type 6 with long term misdiagnosis].
[Two cases of skeletal ciliopathies in one family].
A homozygous frameshift variant in the CILK1 gene causes cranioectodermal dysplasia.
Expanding the genetic spectrum of short rib polydactyly syndrome: Novel DYNC2H1 variants and functional insights.
A novel NEK1 variant disturbs the interaction between the C-terminal fragment of NEK1 and the VDAC1 channel, causing lethal short-rib polydactyly syndrome.
📚 EuropePMC112 artigos no totalmostrando 68
[Clinical characteristics and prenatal diagnosis of a fetus with Short-rib thoracic dysplasia syndrome due to variants of DYNC2H1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics[Clinical analysis of a patient of Short rib-polydactyly syndrome type 6 with long term misdiagnosis].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics[Two cases of skeletal ciliopathies in one family].
Zeitschrift fur Geburtshilfe und NeonatologieA homozygous frameshift variant in the CILK1 gene causes cranioectodermal dysplasia.
European journal of human genetics : EJHGExpanding the genetic spectrum of short rib polydactyly syndrome: Novel DYNC2H1 variants and functional insights.
BonePhenotypic heterogeneity in DYNC2H1-related short-rib thoracic dysplasia: antenatal indicators and postnatal outcomes.
Journal of medical geneticsA novel NEK1 variant disturbs the interaction between the C-terminal fragment of NEK1 and the VDAC1 channel, causing lethal short-rib polydactyly syndrome.
BoneComplete loss of IFT27 function leads to a phenotypic spectrum of fetal lethal ciliopathy associated with altered ciliogenesis.
European journal of human genetics : EJHGA novel compound heterozygous mutation in the DYNC2H1 gene in a Chinese family with Jeune syndrome.
HereditasCompound Heterozygous Variants in the IFT140 Gene Associated with Skeletal Ciliopathies.
Diagnostics (Basel, Switzerland)DYNC2H1 splicing variants causing severe prenatal short-rib polydactyly syndrome and postnatal orofaciodigital syndrome.
Annals of human geneticsBiallelic loss of function variants in FUZ result in an orofaciodigital syndrome.
European journal of human genetics : EJHGA novel variant in IFT122 associated with a severe phenotype of cranioectodermal dysplasia.
Congenital anomaliesLenz-Majewski syndrome and recurrent otitis media: Are they related or not?
European journal of medical geneticsClinical features and genetic analysis of a case series of skeletal ciliopathies in a prenatal setting.
BMC medical genomicsRNA sequencing resolves novel DYNC2H1 variants causing short-rib thoracic dysplasia type 3: Case report.
Molecular genetics & genomic medicineEarly prenatal diagnosis of a recurrent case of short-rib thoracic dysplasia 3 due to compound heterozygosity for variations in the DYNC2H1 gene: an "ultrasound first" approach.
The journal of maternal-fetal & neonatal medicine : the official journal of the European Association of Perinatal Medicine, the Federation of Asia and Oceania Perinatal Societies, the International Society of Perinatal ObstetriciansCharacterization of a novel deep-intronic variant in DYNC2H1 identified by whole-exome sequencing in a patient with a lethal form of a short-rib thoracic dysplasia type III.
Cold Spring Harbor molecular case studiesA case of siblings with juvenile retinitis pigmentosa associated with NEK1 gene variants.
Ophthalmic geneticsDisease-associated mutations in WDR34 lead to diverse impacts on the assembly and function of dynein-2.
Journal of cell scienceWhole exome sequencing, clinical exome or targeted gene panels: what to choose for suspected lethal skeletal dysplasia (short rib thoracic dysplasia type IV).
BMJ case reports[Family analysis of a child with Short-rib polydactyly syndrome type III due to variant of DYNC2H1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsMolecular basis underlying the ciliary defects caused by IFT52 variations found in skeletal ciliopathies.
Molecular biology of the cellCorrigendum: TALPID3/KIAA0586 Regulates Multiple Aspects of Neuromuscular Patterning During Gastrointestinal Development in Animal Models and Human.
Frontiers in molecular neuroscienceCompound heterozygous variants in DYNC2H1 in a foetus with type III short rib-polydactyly syndrome and situs inversus totalis.
BMC medical genomicsTALPID3/KIAA0586 Regulates Multiple Aspects of Neuromuscular Patterning During Gastrointestinal Development in Animal Models and Human.
Frontiers in molecular neuroscienceThe Role of Sonic Hedgehog in Human Holoprosencephaly and Short-Rib Polydactyly Syndromes.
International journal of molecular sciencesTtc30a affects tubulin modifications in a model for ciliary chondrodysplasia with polycystic kidney disease.
Proceedings of the National Academy of Sciences of the United States of AmericaReview: Cytoplasmic dynein motors in photoreceptors.
Molecular visionCiliary Dyneins and Dynein Related Ciliopathies.
CellsRadiological and histopathological features of short rib‑polydactyly syndrome type III and identification of two novel DYNC2H1 variants.
Molecular medicine reportsClinical insights and molecular study of three foetuses with DYNC2H1 gene mutation causing short rib thoracic dystrophy.
Clinical geneticsSRPS associated protein WDR60 regulates the multipolar-to-bipolar transition of migrating neurons during cortical development.
Cell death & diseaseShort rib thoracic dysplasia without polydactyly due to novel variant in IFT172 gene.
Journal of postgraduate medicineWhole-exome sequencing identified two novel mutations of DYNC2LI1 in fetal skeletal ciliopathy.
Molecular genetics & genomic medicineExpanding the phenotypic spectrum of IFT81: Associated ciliopathy syndrome.
American journal of medical genetics. Part AThe splice c.1815G>A variant in KIAA0586 results in a phenotype bridging short-rib-polydactyly and oral-facial-digital syndrome: A case report and literature review.
MedicineShort-rib polydactyly syndrome presenting with recurrent severe first-trimester phenotypes: the utility of exome sequencing in deciphering variants of DYNC2H1 gene.
Journal of obstetrics and gynaecology : the journal of the Institute of Obstetrics and GynaecologyAnalysis of transgenic zebrafish expressing the Lenz-Majewski syndrome gene PTDSS1 in skeletal cell lineages.
F1000ResearchDown-regulated WDR35 contributes to fetal anomaly via regulation of osteogenic differentiation.
GeneMutations in IFT80 cause SRPS Type IV. Report of two families and review.
American journal of medical genetics. Part AEarly prenatal detection of short-rib polydactyly syndrome in a monochorionic diamniotic twin pregnancy.
Congenital anomaliesIFT52 as a Novel Candidate for Ciliopathies Involving Retinal Degeneration.
Investigative ophthalmology & visual scienceCorrigendum to "Identification of a c.544C>T mutation in WDR34 as a deleterious recessive allele of short rib-polydactyly syndrome" [Taiwanese Journal of Obstetrics & Gynecology 56 (2017) 857-862].
Taiwanese journal of obstetrics & gynecologyMutation of FOP/FGFR1OP in mice recapitulates human short rib-polydactyly ciliopathy.
Human molecular geneticsEnriched expression of the ciliopathy gene Ick in cell proliferating regions of adult mice.
Gene expression patterns : GEPPrenatal diagnosis of short-rib polydactyly syndrome type III or short-rib thoracic dysplasia 3 with or without polydactyly (SRTD3) associated with compound heterozygous mutations in DYNC2H1 in a fetus.
Taiwanese journal of obstetrics & gynecologyTargeted gene panel sequencing prenatally detects two novel mutations of DYNC2H1 in a fetus with increased biparietal diameter and polyhydramnios.
Birth defects researchExpanding the phenotype associated with biallelic WDR60 mutations: Siblings with retinal degeneration and polydactyly lacking other features of short rib thoracic dystrophies.
American journal of medical genetics. Part AIdentification of a c.544C>T mutation in WDR34 as a deleterious recessive allele of short rib-polydactyly syndrome.
Taiwanese journal of obstetrics & gynecologyShort rib syndrome Beemer-Langer type, a short history.
American journal of medical genetics. Part AExome sequencing for the differential diagnosis of ciliary chondrodysplasias: Example of a WDR35 mutation case and review of the literature.
European journal of medical geneticsDynein-Driven Retrograde Intraflagellar Transport Is Triphasic in C. elegans Sensory Cilia.
Current biology : CBMutations in IFT-A satellite core component genes IFT43 and IFT121 produce short rib polydactyly syndrome with distinctive campomelia.
CiliaBeemer-Langer syndrome is a ciliopathy due to biallelic mutations in IFT122.
American journal of medical genetics. Part AMutations in DYNC2H1, the cytoplasmic dynein 2, heavy chain 1 motor protein gene, cause short-rib polydactyly type I, Saldino-Noonan type.
Clinical geneticsDestabilization of the IFT-B cilia core complex due to mutations in IFT81 causes a Spectrum of Short-Rib Polydactyly Syndrome.
Scientific reportsIFT52 mutations destabilize anterograde complex assembly, disrupt ciliogenesis and result in short rib polydactyly syndrome.
Human molecular geneticsAn inactivating mutation in intestinal cell kinase, ICK, impairs hedgehog signalling and causes short rib-polydactyly syndrome.
Human molecular geneticsIdentification of novel DYNC2H1 mutations associated with short rib-polydactyly syndrome type III using next-generation panel sequencing.
Genetics and molecular research : GMRMechanism of pancreatic and liver malformations in human fetuses with short-rib polydactyly syndrome.
Birth defects research. Part A, Clinical and molecular teratologyA relatively mild skeletal ciliopathy phenotype consistent with cranioectodermal dysplasia is associated with a homozygous nonsynonymous mutation in WDR35.
American journal of medical genetics. Part AMutations in human homologue of chicken talpid3 gene (KIAA0586) cause a hybrid ciliopathy with overlapping features of Jeune and Joubert syndromes.
Journal of medical geneticsMutations in KIAA0586 Cause Lethal Ciliopathies Ranging from a Hydrolethalus Phenotype to Short-Rib Polydactyly Syndrome.
American journal of human geneticsA Rare Cause of Persistent Pulmonary Hypertension Resistant to Therapy in The Newborn: Short-Rib Polydactyly Syndrome.
Case reports in pulmonologyMutations in DYNC2LI1 disrupt cilia function and cause short rib polydactyly syndrome.
Nature communicationsTargeted next-generation sequencing identifies novel compound heterozygous mutations of DYNC2H1 in a fetus with short rib-polydactyly syndrome, type III.
Clinica chimica acta; international journal of clinical chemistryTctex1d2 associates with short-rib polydactyly syndrome proteins and is required for ciliogenesis.
Cell cycle (Georgetown, Tex.)Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de costela curta-polidactilia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de costela curta-polidactilia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- A homozygous frameshift variant in the CILK1 gene causes cranioectodermal dysplasia.
- Complete loss of IFT27 function leads to a phenotypic spectrum of fetal lethal ciliopathy associated with altered ciliogenesis.
- [Clinical characteristics and prenatal diagnosis of a fetus with Short-rib thoracic dysplasia syndrome due to variants of DYNC2H1 gene].Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics· 2025· PMID 41645379mais citado
- [Clinical analysis of a patient of Short rib-polydactyly syndrome type 6 with long term misdiagnosis].Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical genetics· 2025· PMID 41230591mais citado
- [Two cases of skeletal ciliopathies in one family].
- Expanding the genetic spectrum of short rib polydactyly syndrome: Novel DYNC2H1 variants and functional insights.
- A novel NEK1 variant disturbs the interaction between the C-terminal fragment of NEK1 and the VDAC1 channel, causing lethal short-rib polydactyly syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:1505(Orphanet)
- MONDO:0015461(MONDO)
- GARD:18726(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4420146(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar