Anomalia congênita múltipla caracterizada por insuficiência pancreática exócrina, hipoplasia/aplasia das asas nasais, hipodontia, perda auditiva neurossensorial, retardo de crescimento, malformações anais e urogenitais e deficiência intelectual variável.
Introdução
O que você precisa saber de cara
Anomalia congênita múltipla caracterizada por insuficiência pancreática exócrina, hipoplasia/aplasia das asas nasais, hipodontia, perda auditiva neurossensorial, retardo de crescimento, malformações anais e urogenitais e deficiência intelectual variável.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 39 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 92 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisE3 ubiquitin-protein ligase which is a component of the N-end rule pathway (PubMed:15548684, PubMed:16311597, PubMed:18162545, PubMed:20835242, PubMed:28392261). Recognizes and binds proteins bearing specific N-terminal residues that are destabilizing according to the N-end rule, leading to their ubiquitination and subsequent degradation (PubMed:18162545, PubMed:20835242, PubMed:28392261). Recognizes both type-1 and type-2 N-degrons, containing positively charged amino acids (Arg, Lys and His) a
Cytoplasm, cytosol
Johanson-Blizzard syndrome
This disorder includes congenital exocrine pancreatic insufficiency, multiple malformations such as nasal wing aplasia, and intellectual disability. Pancreas of individuals with JBS do not express UBR1 and show intrauterine-onset destructive pancreatitis.
Variantes genéticas (ClinVar)
117 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 46 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Johanson-Blizzard
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Three Yemeni Siblings With Johanson-Blizzard Syndrome: A Case Report and Literature Review.
Johanson-Blizzard syndrome (JBS), also known as UBR1-related disorder, is a very rare autosomal recessive disorder caused by pathogenic variants in the UBR1 gene and characterized by significant phenotypic variability. The condition is known to be mainly characterized by craniofacial abnormalities, exocrine pancreatic insufficiency, growth retardation, and sensorineural hearing loss. We describe three affected siblings from a consanguineous Yemeni family with JBS. Two brothers suffered from profound symptoms resulting in infant death, which included failure to thrive, exocrine pancreatic dysfunction, anemia, hypoalbuminemia, aplasia cutis congenita, and cardiomyopathy, which was only present in one sibling. The third sibling, who is still alive, is a one-year-old girl who presented with vomiting, diarrhea, failure to thrive, and marked facial dysmorphic features, including hypoplastic alae nasi, a beaked nose, brachycephaly, and a fifth-finger anomaly, without significant visceral malformations. Genome analysis of the affected sibling revealed a homozygous missense mutation in the UBR1 gene, following the American College of Medical Genetics and Genomics (ACMG) guidelines. Moreover, the familial form, consanguinity, and typical presentation are highly suggestive of a diagnosis of JBS. The current case report draws attention to the significant variability of JBS within families and, once again, emphasizes the need for precise clinical assessment in order to make a diagnosis, especially when molecular testing might be equivocal in resource-poor environments. Early multidisciplinary supportive care and genetic counseling are pivotal for optimizing patient survival and minimizing the rate of recurrence within affected kindreds. A narrative review of the literature was conducted to contextualize the clinical findings and highlight intrafamilial phenotypic variability.
Variable Expressivity in Johanson-Blizzard Syndrome: A Case With Severe Manifestations and a Review of the Literature.
Johanson-Blizzard syndrome (JBS) is an exceedingly rare, autosomal recessively inherited disorder. It affects both males and females equally. Exocrine pancreatic insufficiency is the most common finding of the syndrome. The clinical presentation varies significantly among cases. A 6-month-old infant was referred due to persistent diarrhea and failure to gain weight. The diagnosis of JBS was made based on family history and medical investigations; pancreatic enzymes (Pancreatin) were the first line of therapy besides the fluids, blood transfusions, and vitamins. The exact cause of death remains unclear. This case highlights the severe systemic nature of JBS. Early recognition and comprehensive management are crucial for improving outcomes.
Glutamate metabolism disruption in Johanson-Blizzard syndrome: Insights from C. elegans ubr-1 model.
The Johanson-Blizzard syndrome (JBS) is a complex autosomal recessive disorder that manifests through a spectrum of symptoms, with deficiencies in the ubiquitin-protein ligase E3 component N-recognin 1 (UBR-1) at its genetic core. Despite its clinical significance, the molecular intricacies of UBR-1's role in JBS remain largely elusive, presenting a formidable challenge in devising targeted treatments. The nematode Caenorhabditis elegans, with its genetic tractability and conservation of fundamental biological mechanisms, emerges as an invaluable model for unravelling the molecular underpinnings of JBS. This review integrates the latest discoveries from C. elegans studies, shedding light on UBR-1's multiple functions: its regulatory impact on cellular pathways and, particularly, its crucial involvement in glutamate metabolism. By assessing the contributions of these studies to our understanding of JBS, this review highlights the potential significance of glutamate metabolic dysfunction in JBS pathogenesis.
Heterozygous UBR5 variants result in a neurodevelopmental syndrome with developmental delay, autism, and intellectual disability.
E3 ubiquitin ligases have been linked to developmental diseases including autism, Angelman syndrome (UBE3A), and Johanson-Blizzard syndrome (JBS) (UBR1). Here, we report variants in the E3 ligase UBR5 in 29 individuals presenting with a neurodevelopmental syndrome that includes developmental delay, autism, intellectual disability, epilepsy, movement disorders, and/or genital anomalies. Their phenotype is distinct from JBS due to the absence of exocrine pancreatic insufficiency and the presence of autism, epilepsy, and, in some probands, a movement disorder. E3 ubiquitin ligases are responsible for transferring ubiquitin to substrate proteins to regulate a variety of cellular functions, including protein degradation, protein-protein interactions, and protein localization. Knocking out ubr-5 in C. elegans resulted in a lower movement score compared to the wild type, supporting a role for UBR5 in neurodevelopment. Using an in vitro autoubiquitination assay and confocal microscopy for the human protein, we found decreased ubiquitination activity and altered cellular localization in several variants found in our cohort compared to the wild type. In conclusion, we found that variants in UBR5 cause a neurodevelopmental syndrome that can be associated with a movement disorder, reinforcing the role of the UBR protein family in a neurodevelopmental disease that differs from previously described ubiquitin-ligase-related syndromes. We also provide evidence for the pathogenic potential loss of UBR5 function with functional experiments in C. elegans and in vitro ubiquitination assays.
UBR-1 enzyme network regulates glutamate homeostasis to affect organismal behavior and developmental viability.
Johanson-Blizzard Syndrome (JBS) is an autosomal recessive spectrum disorder associated with the UBR-1 ubiquitin ligase that features developmental delay including motor abnormalities. Here, we demonstrate that C. elegans UBR-1 regulates high-intensity locomotor behavior and developmental viability via both ubiquitin ligase and scaffolding mechanisms. Super-resolution imaging with CRISPR-engineered UBR-1 and genetic results demonstrated that UBR-1 is expressed and functions in the nervous system including in pre-motor interneurons. To decipher mechanisms of UBR-1 function, we deployed CRISPR-based proteomics using C. elegans which identified a cadre of glutamate metabolic enzymes physically associated with UBR-1 including GLN-3, GOT-2.2, GFAT-1 and GDH-1. Similar to UBR-1, all four glutamate enzymes are genetically linked to human developmental and neurological deficits. Proteomics, multi-gene interaction studies, and pharmacological findings indicated that UBR-1, GLN-3 and GOT-2.2 form a signaling axis that regulates glutamate homeostasis. Developmentally, UBR-1 is expressed in embryos and functions with GLN-3 to regulate viability. Overall, our results suggest UBR-1 is an enzyme hub in a GOT-2.2/UBR-1/GLN-3 axis that maintains glutamate homeostasis required for efficient locomotion and organismal viability. Given the prominent role of glutamate within and outside the nervous system, the UBR-1 glutamate homeostatic network we have identified could contribute to JBS etiology.
Publicações recentes
Three Yemeni Siblings With Johanson-Blizzard Syndrome: A Case Report and Literature Review.
UBR-1 enzyme network regulates glutamate homeostasis to affect organismal behavior and developmental viability.
Variable Expressivity in Johanson-Blizzard Syndrome: A Case With Severe Manifestations and a Review of the Literature.
Vanishing pancreas: CT and MRI features and imaging diagnostic strategies.
Two de novo UBR1 variants in trans as a cause of Johanson-Blizzard syndrome.
📚 EuropePMC90 artigos no totalmostrando 37
Three Yemeni Siblings With Johanson-Blizzard Syndrome: A Case Report and Literature Review.
CureusUBR-1 enzyme network regulates glutamate homeostasis to affect organismal behavior and developmental viability.
bioRxiv : the preprint server for biologyVariable Expressivity in Johanson-Blizzard Syndrome: A Case With Severe Manifestations and a Review of the Literature.
JGH open : an open access journal of gastroenterology and hepatologyVanishing pancreas: CT and MRI features and imaging diagnostic strategies.
Insights into imagingTwo de novo UBR1 variants in trans as a cause of Johanson-Blizzard syndrome.
Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, CzechoslovakiaGlutamate metabolism disruption in Johanson-Blizzard syndrome: Insights from C. elegans ubr-1 model.
Journal of biosciencesHeterozygous UBR5 variants result in a neurodevelopmental syndrome with developmental delay, autism, and intellectual disability.
American journal of human geneticsJohanson-Blizzard syndrome with cystic dilation of the cochlea and hypoplastic modiolus: a case report.
Pediatric radiologyEstablishment of a human induced pluripotent stem cell line (SDQLCHi079-A) from a patient with Johanson-Blizzard syndrome carrying heterozygous mutation in UBR1 gene.
Stem cell researchJohanson-Blizzard syndrome caused by novel UBR1 mutation in four Saudi patients.
JPGN reportsJohanson-Blizzard Syndrome: A Case Report From Bahrain With a Literature Review.
CureusUBR-1 ubiquitin ligase regulates the balance between GABAergic and glutamatergic signaling.
EMBO reports[Analysis of a child with Johanson-Blizzard syndrome due to novel compound heterozygous variants of UBR1 gene].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCongenital etiologies of exocrine pancreatic insufficiency.
Frontiers in pediatricsEvaluation of Fundus Blood Flow Perfusion in Patients with Diabetic Retinopathy after PPV with Fundus Color Doppler Based on Big Data Mining.
Journal of healthcare engineering50 Years Ago in TheJournalofPediatrics: Molecular Diagnostics Determine Underlying Genetic Etiologies for Well-Described Clinical Syndromes.
The Journal of pediatricsAudiological Profiling and Rehabilitation Outcomes in a Child With Johanson-Blizzard Syndrome.
Journal of audiology & otologyPerformance of Children With Johanson-Blizzard Syndrome After Cochlear Implantation.
CureusLoss of protein quality control gene UBR1 sensitizes Saccharomyces cerevisiae to the aminoglycoside hygromycin B.
Fine focusBiallelic deletion in a minimal CAPN15 intron in siblings with a recognizable syndrome of congenital malformations and developmental delay.
Clinical geneticsUBR7 functions with UBR5 in the Notch signaling pathway and is involved in a neurodevelopmental syndrome with epilepsy, ptosis, and hypothyroidism.
American journal of human geneticsJohanson-Blizzard's Syndrome with a Novel UBR1 Mutation.
Journal of pediatric geneticsSevere forms of Johanson-Blizzard syndrome caused by two novel compound heterozygous variants in UBR1: Clinical manifestations, imaging findings and molecular genetics.
Pancreatology : official journal of the International Association of Pancreatology (IAP) ... [et al.]Pancreatic Malnutrition in Children.
Pediatric annalsClinical Characteristics and Genetic Causes of Infantile Exocrine Pancreatic Insufficiency in Chinese Patients: Study From a Tertiary Care Center.
PancreasJohanson-Blizzard syndrome with associated urogenital anomalies.
BMJ case reportsA rare cause of pancreatic insufficiency; Johanson Blizzard Syndrome.
JPMA. The Journal of the Pakistan Medical AssociationExpanding the mutational spectrum in Johanson-Blizzard syndrome: identification of whole exon deletions and duplications in the UBR1 gene by multiplex ligation-dependent probe amplification analysis.
Molecular genetics & genomic medicineBilateral cochlear implantation in a child with Johanson Blizzard Syndrome.
International journal of pediatric otorhinolaryngologyBound Waters Mediate Binding of Diverse Substrates to a Ubiquitin Ligase.
Structure (London, England : 1993)Is a fatty pancreas a banal lesion?
Journal of ultrasonographyA Case with Complete Pancreatic Aplasia Suggestive of Johanson-Blizzard Syndrome.
Journal of clinical and diagnostic research : JCDRJohanson-Blizzard syndrome presenting as chronic diarrhoea.
Tropical gastroenterology : official journal of the Digestive Diseases FoundationPhysiological functions and clinical implications of the N-end rule pathway.
Frontiers of medicineVariants in the UBR1 gene are not associated with chronic pancreatitis in Japan.
Pancreatology : official journal of the International Association of Pancreatology (IAP) ... [et al.]Oblique facial clefts in Johanson-Blizzard syndrome.
American journal of medical genetics. Part ATwo novel UBR1 gene mutations ın a patient with Johanson Blizzard Syndrome: A mild phenotype without mental retardation.
GeneAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Johanson-Blizzard.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Johanson-Blizzard
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Three Yemeni Siblings With Johanson-Blizzard Syndrome: A Case Report and Literature Review.
- Variable Expressivity in Johanson-Blizzard Syndrome: A Case With Severe Manifestations and a Review of the Literature.JGH open : an open access journal of gastroenterology and hepatology· 2025· PMID 40735003mais citado
- Glutamate metabolism disruption in Johanson-Blizzard syndrome: Insights from C. elegans ubr-1 model.
- Heterozygous UBR5 variants result in a neurodevelopmental syndrome with developmental delay, autism, and intellectual disability.
- UBR-1 enzyme network regulates glutamate homeostasis to affect organismal behavior and developmental viability.
- Vanishing pancreas: CT and MRI features and imaging diagnostic strategies.
- Two de novo UBR1 variants in trans as a cause of Johanson-Blizzard syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2315(Orphanet)
- OMIM OMIM:243800(OMIM)
- MONDO:0009479(MONDO)
- GARD:80(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1699007(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
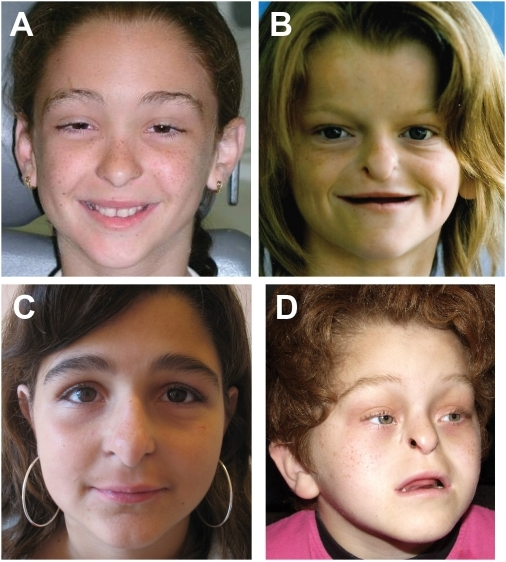
Síndrome Johanson-Blizzard
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata