A síndrome de Antley-Bixler é uma condição raríssima caracterizada pelo fechamento precoce dos ossos do crânio, desenvolvimento insuficiente da parte central do rosto, união dos ossos do antebraço e do braço, fêmur curvado e limitação dos movimentos nas articulações.
Introdução
O que você precisa saber de cara
A síndrome de Antley-Bixler é uma condição raríssima caracterizada pelo fechamento precoce dos ossos do crânio, desenvolvimento insuficiente da parte central do rosto, união dos ossos do antebraço e do braço, fêmur curvado e limitação dos movimentos nas articulações.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 56 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 106 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
2 genes identificados com associação a esta condição. Padrão de herança: Autosomal dominant, Autosomal recessive.
Tyrosine-protein kinase that acts as a cell-surface receptor for fibroblast growth factors and plays an essential role in the regulation of cell proliferation, differentiation, migration and apoptosis, and in the regulation of embryonic development. Required for normal embryonic patterning, trophoblast function, limb bud development, lung morphogenesis, osteogenesis and skin development. Plays an essential role in the regulation of osteoblast differentiation, proliferation and apoptosis, and is
Cell membraneGolgi apparatusCytoplasmic vesicleSecreted
Crouzon syndrome
An autosomal dominant syndrome characterized by craniosynostosis, hypertelorism, exophthalmos and external strabismus, parrot-beaked nose, short upper lip, hypoplastic maxilla, and a relative mandibular prognathism.
This enzyme is required for electron transfer from NADP to cytochrome P450 in microsomes. It can also provide electron transfer to heme oxygenase and cytochrome B5
Endoplasmic reticulum membrane
Antley-Bixler syndrome, with genital anomalies and disordered steroidogenesis
A disease characterized by the association of Antley-Bixler syndrome with steroidogenesis defects and abnormal genitalia. Antley-Bixler syndrome is characterized by craniosynostosis, radiohumeral synostosis present from the perinatal period, midface hypoplasia, choanal stenosis or atresia, femoral bowing and multiple joint contractures.
Variantes genéticas (ClinVar)
502 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 182 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
18 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Antley-Bixler
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Humeroradial Synostosis: An Updated Classification and Differential Diagnosis Based on Genetic Aetiology.
Humeroradial synostosis (HRS) is a rare congenital limb malformation, characterised by fusion of the humeral and radial bones, leading to functional disability of the elbow joint. HRS may be reported in familial or sporadic cases and observed either isolated or as part of a syndromic condition. According to an extensive review of the literature, a dozen known conditions may comprise an HRS. The present review aims to propose an updated classification based on molecular pathways (chondrogenesis and osteogenesis; limb development and patterning; genome regulation), combined with a concise overview of the conditions associated with HRS. This knowledge could guide molecular analyses, patient management and genetic counselling. As some cases remain unexplained, further genetic and epidemiological studies are required to evaluate the contribution of genetic and environmental factors in HRS physiopathology.
CYP51A1 in health and disease: from sterol metabolism to regulated cell death.
How do cells precisely coordinate sterol metabolism with survival and death signals in diverse physiological and pathological contexts? This fundamental question has gained increasing attention as accumulating evidence reveals that enzymes traditionally associated with lipid biosynthesis may have unexpected regulatory functions beyond metabolism. Cytochrome P450 family 51 subfamily A member 1 (CYP51A1), a conserved sterol 14α-demethylase essential for cholesterol synthesis, exemplifies this emerging concept. Although well-characterized as an antifungal drug target in microorganisms, the roles of human CYP51A1 in development, cell death regulation, and disease pathogenesis remain underexplored. Recent studies have uncovered that CYP51A1 not only contributes to cholesterol homeostasis but also modulates multiple forms of regulated cell death-including apoptosis, ferroptosis, alkaliptosis, and pyroptosis-via sterol intermediates or cholesterol-independent mechanisms. Moreover, dysregulation of CYP51A1 has been implicated in a wide spectrum of diseases, such as cancer, cataracts, Antley-Bixler syndrome, autoimmune disorders, metabolic liver disease and neurodegeneration. In this review, we provide a comprehensive synthesis of CYP51A1's structure, regulatory networks, and non-canonical functions. We propose a unifying framework in which CYP51A1 integrates metabolic reprogramming and cell fate control, highlighting its potential as a therapeutic target across diverse human diseases.
Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
P450 oxidoreductase deficiency (PORD) affects cytochrome enzyme activities, causing various symptoms, such as adrenal insufficiency, disorders of sex development and skeletal malformations. This study aims to elucidate the clinical manifestations, genotype characteristics, diagnosis and management of 46,XX karyotype patients with PORD in China. A retrospective study included twelve 46,XX PORD patients in a Chinese tertiary medical center from 2004 to 2024. The patients' clinical characteristics were summarized based on manifestations, hormone profiles, and responses to treatments. The age of first visit was 7-31 years. Except for one young girl presenting with ambiguous genitalia since born, 11 patients presented with either abnormal menses or multiple ovarian cysts. Six patients showed masculinization of their external genitalia, and ten patients showed varying degrees of skeletal deformity. Progesterone was elevated and ovarian reserve was poor in all patients. The most frequent POR variant, c.1370G > A, located in exon 11 occurred in 11/12 patients with an allele frequency of 87.5% (21/24). Two novel nonsense mutations, c.1684dupG and c.2040dupC, were identified and assessed as pathogenic and likely pathogenic by ACMG, respectively. The c.1370G > A might be a dominant mutation type of POR in China. Female patients with PORD have a vulnerable ovarian reserve, and their ovarian macrocysts can be managed conservatively for fertility preservation. This study specifically focuses on PORD in 46,XX Chinese individuals, which implies its genetic causes with novel genetic findings and summarizes the puzzling spectrum of clinical manifestations.
Antley-Bixler syndrome: a case report on virtual planning for monobloc distraction osteogenesis and a surgical intervention narrative review.
Antley-Bixler syndrome (ABS) is an extremely rare genetic disorder characterized by distinct features such as trapezoidal face craniosynostosis, midface hypoplasia with exorbitism, depressed nasal bridge, chonal atresia, radio-humeral synostosis, joint contractures and arachnodactyly. The aim of our study is to: (i) Comprehensively analyse available literature on ABS and report on the surgical management as well as treatment outcomes. (ii) Describe a case of ABS that has been successfully treated with monobloc distraction osteogenesis via virtual surgical planning. A PUBMED search was performed in June 2024. The search was based on a general search string limited to "Antley-Bixler". Inclusion criteria included systematic reviews preferably with meta-analysis, randomized controlled trials, cohort studies and case reports that were reported in English. Papers that focussed on surgical techniques were only present in the form of case reports and case series, out of which only 8 papers that fulfilled the inclusion criteria were included. Surgical techniques that were reported on included fronto-orbital advancement, midfacial advancement, distraction osteogenesis and others. The reported patients also underwent several other surgical procedures for either functional or aesthetic purposes. All the patients reported a good outcome from their surgical interventions. Monobloc distraction osteogenesis is one of the most reliable and predictable procedures that may solve various problems associated with complex craniofacial deformities such as in ABS. This can be seen from the favourable outcome reported in several studies. However, comprehensive pre-surgical virtual planning and multidisciplinary care contributes greatly to favourable functional outcomes.
Diagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
Cytochrome P450 oxidoreductase deficiency (PORD) is a rare form of congenital adrenal hyperplasia that can manifest with skeletal malformations, ambiguous genitalia, and menstrual disorders caused by cytochrome P450 oxidoreductase (POR) mutations affecting electron transfer to all microsomal cytochrome P450 and some non-P450 enzymes involved in cholesterol, sterol, and drug metabolism. With the advancement of molecular biology and medical genetics, increasing numbers of PORD cases were reported, and the clinical spectrum of PORD was extended with studies on underlying mechanisms of phenotype-genotype correlations and optimum treatment. However, diagnostic challenges and management dilemma still exists because of unawareness of the condition, the overlapping manifestations with other disorders, and no clear guidelines for treatment. Delayed diagnosis and management may result in improper sex assignment, loss of reproductive capacity because of surgical removal of ruptured ovarian macro-cysts, and life-threatening conditions such as airway obstruction and adrenal crisis. The clinical outcomes and prognosis, which are influenced by specific POR mutations, the presence of additional genetic or environmental factors, and management, include early death due to developmental malformations or adrenal crisis, bilateral oophorectomies after spontaneous rupture of ovarian macro-cysts, genital ambiguity, abnormal pubertal development, and nearly normal phenotype with successful pregnancy outcomes by assisted reproduction. Thus, timely diagnosis including prenatal diagnosis with invasive and non-invasive techniques and appropriate management is essential to improve patients' outcomes. However, even in cases with conclusive diagnosis, comprehensive assessment is needed to avoid severe complications, such as chromosomal test to help sex assignment and evaluation of adrenal function to detect partial adrenal insufficiency. In recent years, it has been noted that proper hormone replacement therapy can lead to decrease or resolve of ovarian macro-cysts, and healthy babies can be delivered by in vitro fertilization and frozen embryo transfer following adequate control of multiple hormonal imbalances. Treatment may be complicated with adverse effects on drug metabolism caused by POR mutations. Unique challenges occur in female PORD patients such as ovarian macro-cysts prone to spontaneous rupture, masculinized genitalia without progression after birth, more frequently affected pubertal development, and impaired fertility. Thus, this review focuses only on 46, XX PORD patients to summarize the potential molecular pathogenesis, differential diagnosis of classic and non-classic PORD, and tailoring therapy to maintain health, avoid severe complications, and promote fertility.
Publicações recentes
Humeroradial Synostosis: An Updated Classification and Differential Diagnosis Based on Genetic Aetiology.
CYP51A1 in health and disease: from sterol metabolism to regulated cell death.
Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
Antley-Bixler syndrome: a case report on virtual planning for monobloc distraction osteogenesis and a surgical intervention narrative review.
A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
📚 EuropePMC94 artigos no totalmostrando 51
Humeroradial Synostosis: An Updated Classification and Differential Diagnosis Based on Genetic Aetiology.
Clinical geneticsCYP51A1 in health and disease: from sterol metabolism to regulated cell death.
Cell death discoveryClinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
Reproductive sciences (Thousand Oaks, Calif.)Antley-Bixler syndrome: a case report on virtual planning for monobloc distraction osteogenesis and a surgical intervention narrative review.
Journal of stomatology, oral and maxillofacial surgeryDiagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
Frontiers in endocrinologyCongenital adrenal hyperplasia due to P450 oxidoreductase deficiency.
Frontiers in endocrinologyIn Silico Analysis of PORD Mutations on the 3D Structure of P450 Oxidoreductase.
Molecules (Basel, Switzerland)A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
American journal of human geneticsThe first adult case of cytochrome P450 oxidoreductase deficiency with sufficient semen volume and sperm concentration.
Congenital anomaliesClassic and current concepts in adrenal steroidogenesis: a reappraisal.
Archives of endocrinology and metabolismAntley-Bixler syndrome arising from compound heterozygotes in the P450 oxidoreductase gene: a case report.
Translational pediatricsTwo cases of cytochrome P450 oxidoreductase deficiency with severe scoliosis and surgery requirement.
Congenital anomaliesEstablishment of an induced pluripotent stem cell line from a Antley-Bixler Syndrome (ABS) patient with the homozygote mutation p.R457H (c.1370G>A) in POR gene.
Stem cell researchLow-birth-weight infant with Antley-Bixler syndrome-like phenotype caused by POR mutation: a rare case report.
European review for medical and pharmacological sciencesCytochrome P450 oxidoreductase deficiency caused by a novel mutation in the POR gene in two siblings: case report and literature review.
Hormones (Athens, Greece)Next-Generation Sequencing Revealed Disease-Causing Variants in Two Genes in a Patient With Combined Features of Spherocytosis and Antley-Bixler Syndrome With Genital Anomalies and Disordered Steroidogenesis.
Frontiers in geneticsIdentifying the Misshapen Head: Craniosynostosis and Related Disorders.
Pediatrics[Clinical and genetic analysis of a pedigree affected with cytochrome P450 oxidoreductase deficiency].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsClinical and genetic analysis of cytochrome P450 oxidoreductase (POR) deficiency in a female and the analysis of a novel POR intron mutation causing alternative mRNA splicing : Overall analysis of a female with POR deficiency.
Journal of assisted reproduction and geneticsClinical, endocrinological, and molecular features of four Korean cases of cytochrome P450 oxidoreductase deficiency.
Annals of pediatric endocrinology & metabolismClinical characteristics of cytochrome P450 oxidoreductase deficiency: a nationwide survey in Japan.
Endocrine journalNon-classic cytochrome P450 oxidoreductase deficiency strongly linked with menstrual cycle disorders and female infertility as primary manifestations.
Human reproduction (Oxford, England)Novel phenotypes and genotypes in Antley-Bixler syndrome caused by cytochrome P450 oxidoreductase deficiency: based on the first cohort of Chinese children.
Orphanet journal of rare diseasesP450 Oxidoreductase Deficiency: A Systematic Review and Meta-analysis of Genotypes, Phenotypes, and Their Relationships.
The Journal of clinical endocrinology and metabolismAlternative pathway androgen biosynthesis and human fetal female virilization.
Proceedings of the National Academy of Sciences of the United States of America[A case of Antley-Bixler syndrome caused by novel POR mutations].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsFurther Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes.
GenesPerinasal Osteotomy With Distraction Osteogenesis for a Mild Syndromic Craniosynostosis.
The Journal of craniofacial surgeryCompound heterozygous variants in POR gene identified by whole-exome sequencing in a Chinese pedigree with cytochrome P450 oxidoreductase deficiency.
Pediatric investigationRETRACTED ARTICLE: Fetal methotrexate syndrome and Antley-Bixler syndrome should not be confused.
Pediatric radiology[Advance in clinical research on Antley-Bixler syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsLongitudinal serum and urine steroid metabolite profiling in a 46,XY infant with prenatally identified POR deficiency.
The Journal of steroid biochemistry and molecular biologyIn vitro fertilization-frozen embryo transfer in a patient with cytochrome P450 oxidoreductase deficiency: a case report.
Gynecological endocrinology : the official journal of the International Society of Gynecological EndocrinologyA Case of Antley-Bixler Syndrome With a Novel Likely Pathogenic Variant (c.529G>C) in the POR Gene.
Annals of laboratory medicineB3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation.
Genetics in medicine : official journal of the American College of Medical GeneticsTracheal Cartilaginous Sleeve in Syndromic Craniosynostosis: An Underrecognized Source of Significant Morbidity and Mortality.
The Journal of craniofacial surgeryCytochrome P450 oxidoreductase deficiency caused by R457H mutation in POR gene in Chinese: case report and literature review.
Journal of ovarian researchLong-term follow-up of a female with congenital adrenal hyperplasia due to P450-oxidoreductase deficiency.
Archives of endocrinology and metabolismP450 Oxidoreductase Deficiency: Loss of Activity Caused by Protein Instability From a Novel L374H Mutation.
The Journal of clinical endocrinology and metabolismInstability of the Human Cytochrome P450 Reductase A287P Variant Is the Major Contributor to Its Antley-Bixler Syndrome-like Phenotype.
The Journal of biological chemistryBiallelic mutations in CYP26B1: A differential diagnosis for Pfeiffer and Antley-Bixler syndromes.
American journal of medical genetics. Part ADelayed diagnosis of disorder of sex development (DSD) due to P450 oxidoreductase (POR) deficiency.
Hormones (Athens, Greece)Genetic Syndromes Associated with Craniosynostosis.
Journal of Korean Neurosurgical SocietyP450 Oxidoreductase deficiency: Analysis of mutations and polymorphisms.
The Journal of steroid biochemistry and molecular biologyCompound heterozygosity of a paternal submicroscopic deletion and a maternal missense mutation in POR gene: Antley-bixler syndrome phenotype in three sibling fetuses.
Birth defects research. Part A, Clinical and molecular teratologyMultidisciplinary Treatment of Antley-Bixler Syndrome.
The Cleft palate-craniofacial journal : official publication of the American Cleft Palate-Craniofacial AssociationP450 oxidoreductase deficiency with maternal virilization during pregnancy.
Clinical and experimental obstetrics & gynecologyPrenatal Diagnosis of Antley-Bixler Syndrome and POR Deficiency.
The American journal of case reportsGross Motor Function of a Child With Antley-Bixler Syndrome.
Pediatric physical therapy : the official publication of the Section on Pediatrics of the American Physical Therapy AssociationFibroblast growth factor receptor signaling in kidney and lower urinary tract development.
Pediatric nephrology (Berlin, Germany)Radiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis.
Pediatric radiologyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Antley-Bixler.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Antley-Bixler
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Humeroradial Synostosis: An Updated Classification and Differential Diagnosis Based on Genetic Aetiology.
- CYP51A1 in health and disease: from sterol metabolism to regulated cell death.
- Clinical Characteristics and Molecular Aetiology of Cytochrome P450 Oxidoreductase Deficiency Diagnosed in 46,XX Patients.
- Antley-Bixler syndrome: a case report on virtual planning for monobloc distraction osteogenesis and a surgical intervention narrative review.
- Diagnostic challenges and management advances in cytochrome P450 oxidoreductase deficiency, a rare form of congenital adrenal hyperplasia, with 46, XX karyotype.
- A spectrum of recessiveness among Mendelian disease variants in UK Biobank.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:83(Orphanet)
- MONDO:0008803(MONDO)
- GARD:5826(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q585011(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
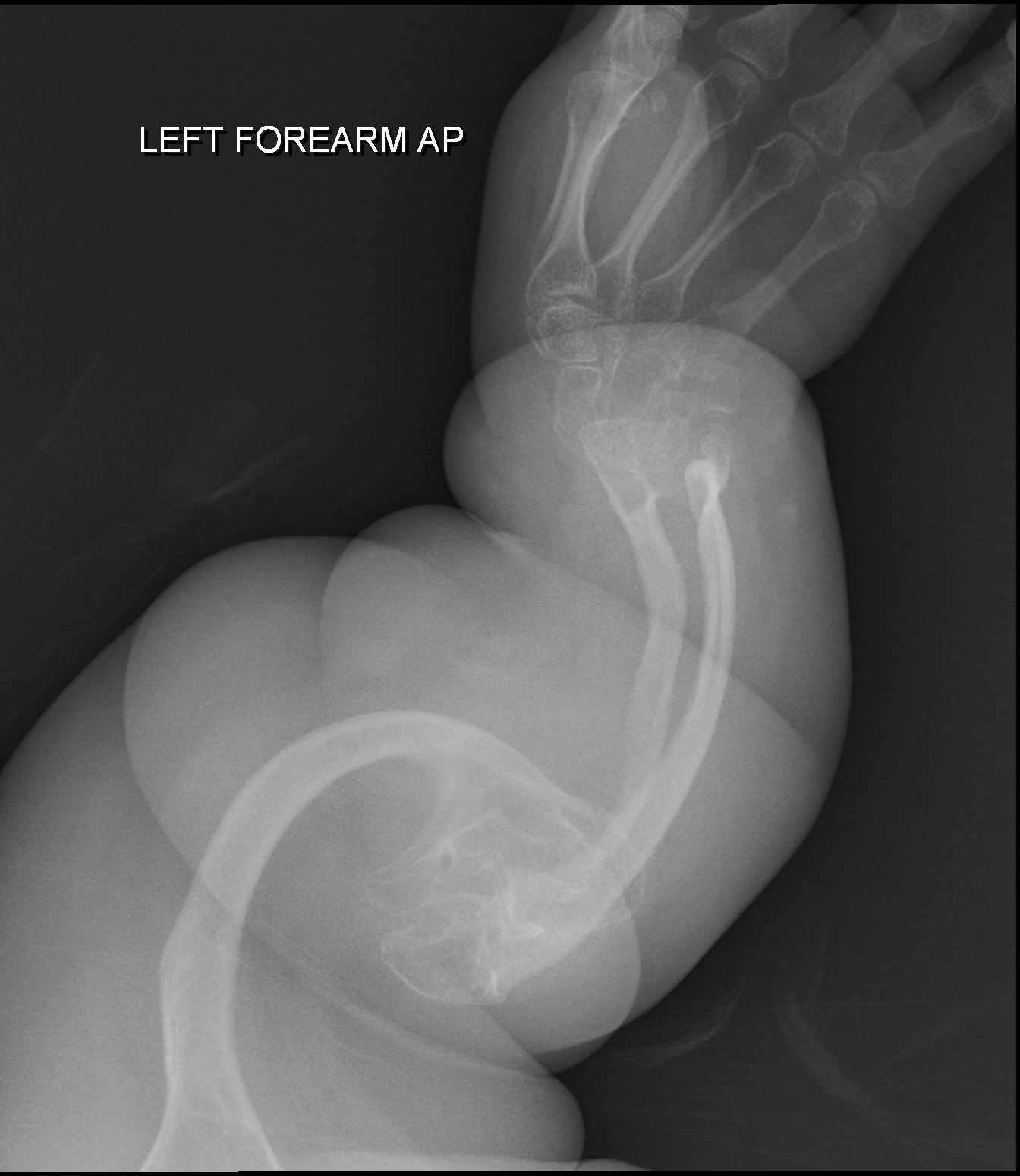
Síndrome Antley-Bixler
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata