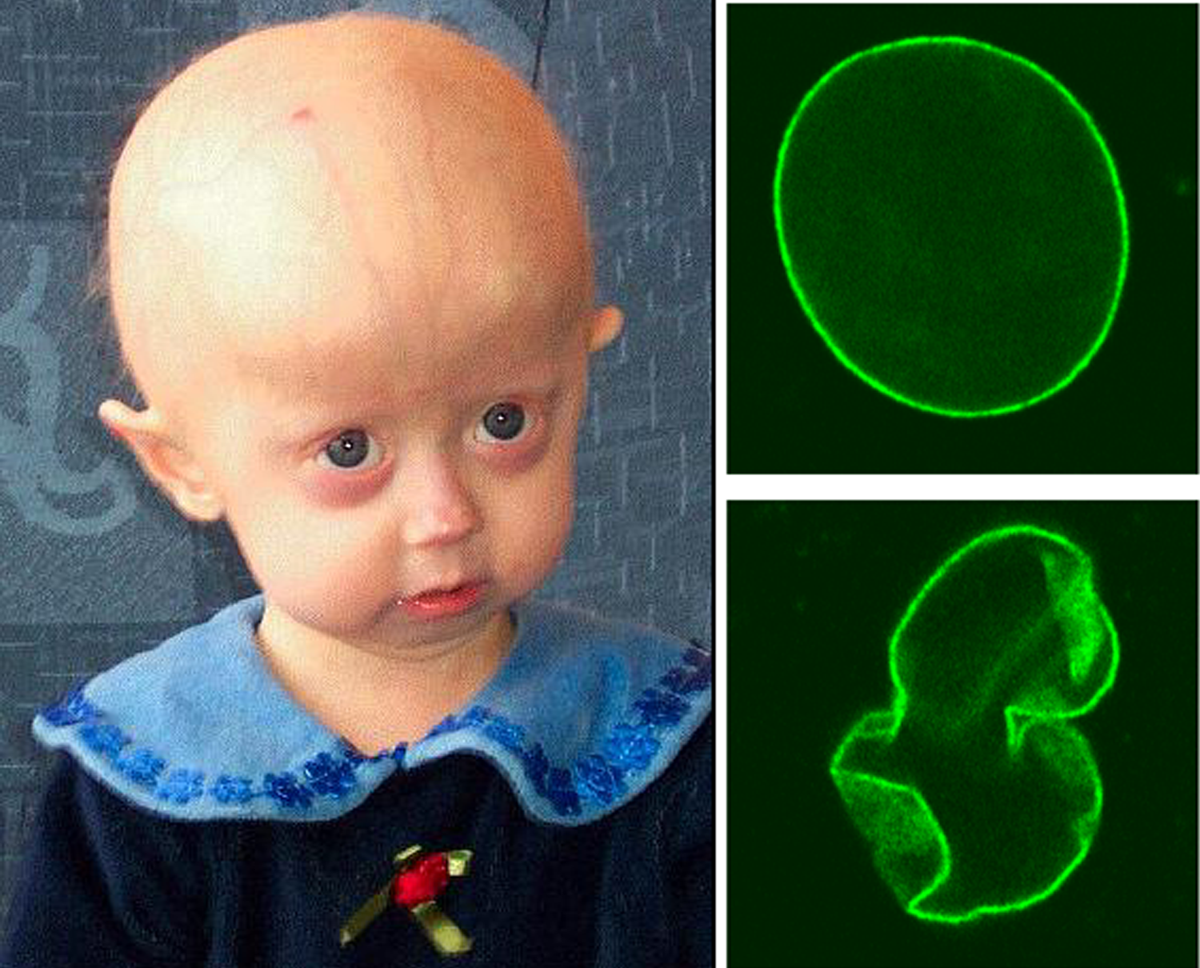
Síndrome rara de envelhecimento prematuro caracterizada por retardo de crescimento pré e pós-natal, aparência congênita de envelhecimento prematuro com dismorfismo craniofacial distinto (cavária larga com fontanela anterior grande e aberta e sutura metópica larga, testa larga, face pequena, micrognatia), gordura subcutânea acentuadamente diminuída, cútis laxa e pele enrugada, sem atraso no desenvolvimento psicomotor. Cabelos escassos e quebradiços, unhas hipoplásicas e dentição anormal e atrasada, bem como falanges distais hipoplásicas, hérnia umbilical e anomalias oculares (miopia/hipermetropia, estrabismo), também estão comumente associados.
Introdução
O que você precisa saber de cara
Síndrome rara de envelhecimento prematuro caracterizada por retardo de crescimento pré e pós-natal, aparência congênita de envelhecimento prematuro com dismorfismo craniofacial distinto (cavária larga com fontanela anterior grande e aberta e sutura metópica larga, testa larga, face pequena, micrognatia), gordura subcutânea acentuadamente diminuída, cútis laxa e pele enrugada, sem atraso no desenvolvimento psicomotor. Cabelos escassos e quebradiços, unhas hipoplásicas e dentição anormal e atrasada, bem como falanges distais hipoplásicas, hérnia umbilical e anomalias oculares (miopia/hipermetropia, estrabismo), também estão comumente associados.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 40 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 122 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal dominant.
Electroneutral antiporter that mediates the transport of adenyl nucleotides through the inner mitochondrial membrane. Originally identified as an ATP-magnesium/inorganic phosphate antiporter, it also acts as a broad specificity adenyl nucleotide antiporter. By regulating the mitochondrial matrix adenyl nucleotide pool, could adapt to changing cellular energetic demands and indirectly regulate adenyl nucleotide-dependent metabolic pathways (PubMed:15123600, PubMed:22015608). In vitro, a low activ
Mitochondrion inner membrane
Fontaine progeroid syndrome
An autosomal dominant progeroid disorder characterized by prenatal and postnatal growth retardation, decreased subcutaneous fat tissue, wrinkled skin, an aged appearance since birth, an abnormal scalp hair pattern, sparse hair, hypoplastic distal phalanges with hypoplastic nails, a widely open anterior fontanel, facial dysmorphisms, and craniosynostosis. Early death is observed in some patients.
Variantes genéticas (ClinVar)
27 variantes patogênicas registradas no ClinVar.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome progeroide, tipo Petty
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Performance evaluation of CMIP6 climate models for selecting a suitable GCM for future precipitation at different places of Tamil Nadu.
Climate change refers to long-term variations in climate parameters. Future climate information can be projected using a GCM (General Circulation Model). Identifying a particular GCM is crucial for climate impact studies. Researchers are perplexed about selecting a suitable GCM for downscaling to predict future climate parameters. Recent updates to CMIP6 global climate models have included shared socioeconomic pathways based on the IPCC (Intergovernmental Panel on Climate Change) Sixth Assessment Report (AR6). The performance of 24 CMIP6 GCMs in precipitation with a multi-model ensemble filter was compared to IMD (India Meteorological Department) 0.25 × 0.25 degrees rainfall data in Tamil Nadu. The performance was evaluated with the help of Compromise Programming (CP), which involves metrics such as R2 (Pearson correlation co-efficient), PBIAS (Percentage Bias), NRMSE (Normalized Root Mean Square Error), and NSE (Nash-Sutcliffe Efficiency). The GCM ranking was performed through Compromise programming by comparing the IMD data and GCM data. The results of the CP analyses of the statistical metrics suggest that the suitable GCMs for the North-East monsoon are CESM2 for Chennai, CAN-ESM5 for Vellore, MIROC6 for Salem, BCC-CSM2-MR for Thiruvannamalai, MPI-ESM-1-2-HAM for Erode, MPI-ESM1-2-LR for Tiruppur, MPI-ESM1-2-LR for Trichy, MPI-ESM1-2-LR for Pondicherry, MPI-ESM1-2-LR for Dindigul, CNRM-CM6-HR for Thanjavur, MPI-ESM1-2-LR for Thirunelveli and UKESM1-0-LL for Thoothukudi. The appropriate suitable GCMs for South-West monsoon as CESM2 is appropriate for Chennai, IPSL-CM6A-LR for Vellore, CESM2-WACCM-FV2 for Salem, CAMS-CSM1-0 for Thiruvannamalai, MPI-ESM-1-2-HR for Erode, MPI-ESM-1-2-HR for Tiruppur, EC- EARTH3 for Trichy, EC- EARTH3 for Pondicherry, MPI-ESM-1-2-HR for Dindigul, CESM2-FV2 for Thanjavur, ACCESS-CM2 for Thirunelveli and ACCESS-CM2 for Thoothukudi respectively. This study emphasizes the importance of selecting an appropriate GCM. Selecting a suitable GCM will be useful in climate change impact studies and there by suggesting necessary adaptation and mitigation strategies.
Different GCMs yet similar outcome: predicting the habitat distribution of Shorea robusta C.F. Gaertn. in the Indian Himalayas using CMIP5 and CMIP6 climate models.
Climate change impact on the habitat distribution of umbrella species presents a critical threat to the entire regional ecosystem. This is further perilous if the species is economically important. Sal (Shorea robusta C.F. Gaertn.), a climax forest forming Central Himalayan tree species, is one of the most valuable timber species and provides several ecological services. Sal forests are under threat due to over-exploitation, habitat destruction, and climate change. Sal's poor natural regeneration and its unimodal density-diameter distribution in the region illustrate the peril to its habitat. We, modelled the current as well as future distribution of suitable sal habitats under different climate scenarios using 179 sal occurrence points and 8 bioclimatic environmental variables (non-collinear). The CMIP5-based RCP4.5 and CMIP6-based SSP245 climate models under 2041-2060 and 2061-2080 periods were used to predict the impact of climate change on sal's future potential distribution area. The niche model results predict the mean annual temperature and precipitation seasonality as the most influential sal habitat governing variables in the region. The current high suitability region for sal was 4.36% of the total geographic area, which shows a drastic decline to 1.31% and 0.07% under SSP245 for 2041-60 and 2061-80, respectively. The RCP-based models predicted more severe impact than SSP; however, both RCP and SSP models showed complete loss of high suitability regions and overall shift of species northwards in the Uttarakhand state. We could identify the current and future suitable habitats for conserving sal population through assisted regeneration and management of other regional issues.
Is Gorlin-Chaudhry-Moss syndrome associated with aortopathy?
Gorlin-Chaudhry-Moss syndrome (GCMS) is a rare disorder consisting of craniofacial dysostosis, hypertrichosis, underdeveloped genitalia, and ocular and dental anomalies. Recently, GCMS has been reclassified together with Fontaine syndrome as Fontaine progeroid syndrome (FPS), after a common genetic basis was found. It was previously thought that GCMS/FPS was not associated with aortopathy, but in recent years 3 patients with aortic disease have been described. We describe the fourth case, who is the oldest patient with GCMS/FPS reported in the medical literature: a 45-year-old patient who presented with acute aortic dissection. We therefore recommend screening patients previously diagnosed with GCMS/FPS for aortic pathology to aid early detection and avoid patient presentation in an acute setting.
A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24.
We report the clinical and genetic findings in a 15-year-old Spanish boy presenting prenatal and postnatal growth retardation, reduced subcutaneous adipose tissue, premature skin wrinkling, sparse hair, short distal phalanges with small nails, umbilical hernia, wide anterior fontanel, and normal cognitive and motor development. Exome sequencing uncovered a heterozygous mutation in SLC25A24 (NM_013386: c.650G>A: p.R217H) that encodes for the calcium-binding mitochondrial carrier protein SCaMC-1. This gain-of-function variant has been previously associated with Fontaine syndrome and Gorlin-Chaudhry-Moss syndrome, two entities that show overlapping features, and have been recently subsumed under the name Fontaine progeroid syndrome (FPS; MIM: 612289) in OMIM. Here, we describe the first male patient with genetically confirmed FPS who survives at least until adolescence.
De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction.
Gorlin-Chaudhry-Moss syndrome (GCMS) is a dysmorphic syndrome characterized by coronal craniosynostosis and severe midface hypoplasia, body and facial hypertrichosis, microphthalmia, short stature, and short distal phalanges. Variable lipoatrophy and cutis laxa are the basis for a progeroid appearance. Using exome and genome sequencing, we identified the recurrent de novo mutations c.650G>A (p.Arg217His) and c.649C>T (p.Arg217Cys) in SLC25A24 in five unrelated girls diagnosed with GCMS. Two of the girls had pronounced neonatal progeroid features and were initially diagnosed with Wiedemann-Rautenstrauch syndrome. SLC25A24 encodes a mitochondrial inner membrane ATP-Mg/Pi carrier. In fibroblasts from affected individuals, the mutated SLC25A24 showed normal stability. In contrast to control cells, the probands' cells showed mitochondrial swelling, which was exacerbated upon treatment with hydrogen peroxide (H2O2). The same effect was observed after overexpression of the mutant cDNA. Under normal culture conditions, the mitochondrial membrane potential of the probands' fibroblasts was intact, whereas ATP content in the mitochondrial matrix was lower than that in control cells. However, upon H2O2 exposure, the membrane potential was significantly elevated in cells harboring the mutated SLC25A24. No reduction of mitochondrial DNA copy number was observed. These findings demonstrate that mitochondrial dysfunction with increased sensitivity to oxidative stress is due to the SLC25A24 mutations. Our results suggest that the SLC25A24 mutations induce a gain of pathological function and link mitochondrial ATP-Mg/Pi transport to the development of skeletal and connective tissue.
Publicações recentes
IVNS1ABP mutation drives cellular senescence in newly identified progeroid neuropathy.
The tight bond between Fanconi anemia and aging.
Fontaine progeroid syndrome into early adolescence: a case report.
Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
Growth Hormone Response in a Child With a Homozygous TOMM7 Mutation: Novel Therapeutic Insights.
📚 EuropePMCmostrando 6
Performance evaluation of CMIP6 climate models for selecting a suitable GCM for future precipitation at different places of Tamil Nadu.
Environmental monitoring and assessmentDifferent GCMs yet similar outcome: predicting the habitat distribution of Shorea robusta C.F. Gaertn. in the Indian Himalayas using CMIP5 and CMIP6 climate models.
Environmental monitoring and assessmentIs Gorlin-Chaudhry-Moss syndrome associated with aortopathy?
European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic SurgeryA rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24.
American journal of medical genetics. Part ADe Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction.
American journal of human geneticsAccurate Detection of Dysmorphic Nuclei Using Dynamic Programming and Supervised Classification.
PloS oneAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome progeroide, tipo Petty.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome progeroide, tipo Petty
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Performance evaluation of CMIP6 climate models for selecting a suitable GCM for future precipitation at different places of Tamil Nadu.
- Different GCMs yet similar outcome: predicting the habitat distribution of Shorea robusta C.F. Gaertn. in the Indian Himalayas using CMIP5 and CMIP6 climate models.
- Is Gorlin-Chaudhry-Moss syndrome associated with aortopathy?European journal of cardio-thoracic surgery : official journal of the European Association for Cardio-thoracic Surgery· 2020· PMID 32355952mais citado
- A rare male patient with Fontaine progeroid syndrome caused by p.R217H de novo mutation in SLC25A24.
- De Novo Mutations in SLC25A24 Cause a Craniosynostosis Syndrome with Hypertrichosis, Progeroid Appearance, and Mitochondrial Dysfunction.
- IVNS1ABP mutation drives cellular senescence in newly identified progeroid neuropathy.
- The tight bond between Fanconi anemia and aging.
- Fontaine progeroid syndrome into early adolescence: a case report.
- Novel POLR3A Gene Mutation Results in Wiedemann-Rautenstrauch Syndrome With Striking Cutis Laxa and Myelofibrosis.
- Growth Hormone Response in a Child With a Homozygous TOMM7 Mutation: Novel Therapeutic Insights.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2963(Orphanet)
- OMIM 612289(OMIM)
- MONDO:0012853(MONDO)
- GARD:4497(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55783883(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome progeroide, tipo Petty
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata