A Aspartilglicosaminúria (AGU) é uma doença genética rara, de herança autossômica recessiva (passada dos pais para os filhos). Ela afeta os lisossomos, que são como "centros de reciclagem" das células, causando o acúmulo de substâncias que deveriam ser processadas (uma doença de depósito lisossômico). A condição pertence ao grupo das oligossacaridoses (também chamadas de glicoproteinose).
Introdução
O que você precisa saber de cara
A Aspartilglicosaminúria (AGU) é uma doença genética rara, de herança autossômica recessiva (passada dos pais para os filhos). Ela afeta os lisossomos, que são como "centros de reciclagem" das células, causando o acúmulo de substâncias que deveriam ser processadas (uma doença de depósito lisossômico). A condição pertence ao grupo das oligossacaridoses (também chamadas de glicoproteinose).
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 29 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 78 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisCleaves the GlcNAc-Asn bond which joins oligosaccharides to the peptide of asparagine-linked glycoproteins
Lysosome
Aspartylglucosaminuria
An inborn lysosomal storage disease causing excess accumulation of glycoasparagine in the body tissues and its increased excretion in urine. Clinical features include mild to severe intellectual disability manifesting from the age of two, coarse facial features and mild connective tissue abnormalities.
Variantes genéticas (ClinVar)
208 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 514 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
1 via biológica associada aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Aspartilglicosaminúria
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
7 ensaios clínicos encontrados, 2 ativos.
Publicações mais relevantes
Congenital Dermal Melanocytosis Associated With Aspartylglucosaminuria: Expanding the Dermatological Phenotype of a Rare Oligosaccharidosis.
A 37-Year-Old Man With Intellectual Disability Discovered to Have Aspartylglucosaminuria: Implications for the Diagnosis of Genetic Causes.
The causes of intellectual disability (ID) are varied, with as many as 1,400 causative genes. We attempted to identify the causative gene in a patient with long-standing undiagnosed ID. Although this was an isolated case with no family history, we searched for the causative gene using trio-based whole-exome sequencing (trio-WES), because severe ID is often caused by genetic variations, and inherited metabolic disorders (IMDs) are assumed to be the cause when regression and epilepsy occur. We identified homozygous donor splice-site variants in the AGA gene (aspartylglucosaminidase; NM_000027.4) Chr4(GRCh38):g. 177436275C>A, c.698+1G>T. This gene is implicated in aspartylglucosaminuria (AGU; OMIM #208400) and originated from both of the patient's parents. We confirmed the pathogenicity of the variant by detecting the splicing defect in cDNA from the patient's blood and accumulation of aberrant metabolites in the patient's urine. We discuss how to more readily achieve an accurate diagnosis for patients with undiagnosed intellectual disabilities. Medical practitioners' awareness of the characteristics of the disease leading to clinical suspicion in patients with matching presentations, and the performance of newborn screening when possible, is important for the diagnosis of ID. In addition, the characteristic symptoms and course of the disease give rise to suspicion of IMDs. Given our results, we consider trio-WES to be a powerful method for identifying the causative genes in cases of ID with genetic causes.
Genetically Confirmed Case of Aspartylglycosaminuria (AGU).
Aspartylglucosaminuria is a lysosomal storage disorder characterized by developmental delay, intellectual disability, behavioral manifestations (hyperactivity in young children, anxiety and restlessness in adolescence, and apathy in adulthood), recurrent infections, musculoskeletal features, and characteristic craniofacial features (prominent supraorbital ridges, hypertelorism, periorbital fullness, short nose with broad nasal bridge, thick vermilion of the upper and lower lips, and macroglossia) that become more prominent with age. Additional neurologic manifestations can include seizures, poor balance and coordination, and progressive cerebral atrophy in adulthood. Macrocephaly is common. Musculoskeletal features include lordosis, scoliosis, and arthritis in adolescents and young adults; vertebral dysplasia and/or rib cage abnormalities; and progressive muscle wasting, joint contractures, bursitis, and osteoporosis in adulthood. Skin manifestations (facial seborrhea, rosacea, and angiofibromas), gastrointestinal manifestations, neutropenia, and thrombocytopenia occur in some individuals. The clinical manifestations of aspartylglucosaminuria worsen with age, and adults have progressive psychomotor decline. The diagnosis of aspartylglucosaminuria can be established in a proband with characteristic clinical and laboratory findings by identification of decreased aspartylglucosaminidase enzymatic activity in serum, leukocytes, or fibroblasts and/or biallelic pathogenic variants in AGA by molecular genetic testing. Treatment of manifestations: Developmental and educational services; standardized treatments for seizures, behavioral manifestations, sleep issues, dental manifestations, recurrent infections, scoliosis, joint swelling and mobility problems, osteoporosis, and gastrointestinal manifestations; social work support and care coordination as needed. Surveillance: At each visit, assess for developmental progress, educational needs, seizures, balance and coordination issues, recurrent infections, spine issues, muscle wasting, joint manifestations, chronic diarrhea or constipation, and family needs. Assess behavioral and sleep issues annually or as needed. Dental examination every six months. Assess bone density every five years, or every two years in those treated for osteoporosis. Complete blood count with differential to assess for neutropenia and thrombocytopenia in those with any clinical manifestations of cytopenia. Evaluations of relatives at where risk: It is appropriate to clarify the genetic status of apparently asymptomatic younger at-risk sibs of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of supportive treatments. Aspartylglucosaminuria is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an AGA pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the AGA pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
A new horizon in the phosphorylated sites of AGA: the structural impact of C163S mutation in aspartylglucosaminuria through molecular dynamics simulation.
Aspartylglucosaminuria (AGU) is a lysosomal storage disorder caused by insufficient aspartylglucosaminidase (AGA) activity leading to chronic neurodegeneration. We utilized the PhosphoSitePlus tool to identify the AGA protein's phosphorylation sites. The phosphorylation was induced on the specific residue of the three-dimensional AGA protein, and the structural changes upon phosphorylation were studied via molecular dynamics simulation. Furthermore, the structural behaviour of C163S mutation and C163S mutation with adjacent phosphorylation was investigated. We have examined the structural impact of phosphorylated forms and C163S mutation in AGA. Molecular dynamics simulations (200 ns) exposed patterns of deviation, fluctuation, and change in compactness of Y178 phosphorylated AGA protein (Y178-p), T215 phosphorylated AGA protein (T215-p), T324 phosphorylated AGA protein (T324-p), C163S mutant AGA protein (C163S), and C163S mutation with Y178 phosphorylated AGA protein (C163S-Y178-p). Y178-p, T215-p, and C163S mutation demonstrated an increase in intramolecular hydrogen bonds, leading to greater compactness of the AGA forms. Principle component analysis (PCA) and Gibbs free energy of the phosphorylated/C163S mutation structures exhibit transition in motion/orientation than Wild type (WT). T215-p may be more dominant among these than the other studied phosphorylated forms. It might contribute to hydrolyzing L-asparagine functioning as an asparaginase, thereby regulating neurotransmitter activity. This study revealed structural insights into the phosphorylation of Y178, T215, and T324 in AGA protein. Additionally, it exposed the structural changes of the C163S mutation and C163S-Y178-p of AGA protein. This research will shed light on a better understanding of AGA's phosphorylated mechanism.Communicated by Ramaswamy H. Sarma.
Validation of Aspartylglucosaminidase Activity Assay for Human Serum Samples: Establishment of a Biomarker for Diagnostics and Clinical Studies.
Novel treatment strategies are emerging for rare, genetic diseases, resulting in clinical trials that require adequate biomarkers for the assessment of the treatment effect. For enzyme defects, biomarkers that can be assessed from patient serum, such as enzyme activity, are highly useful, but the activity assays need to be properly validated to ensure a precise, quantitative measurement. Aspartylglucosaminuria (AGU) is a lysosomal storage disorder caused by the deficiency of the lysosomal hydrolase aspartylglucosaminidase (AGA). We have here established and validated a fluorometric AGA activity assay for human serum samples from healthy donors and AGU patients. We show that the validated AGA activity assay is suitable for the assessment of AGA activity in the serum of healthy donors and AGU patients, and it can be used for diagnostics of AGU and, potentially, for following a treatment effect.
Publicações recentes
Congenital Dermal Melanocytosis Associated With Aspartylglucosaminuria: Expanding the Dermatological Phenotype of a Rare Oligosaccharidosis.
A 37-Year-Old Man With Intellectual Disability Discovered to Have Aspartylglucosaminuria: Implications for the Diagnosis of Genetic Causes.
A new horizon in the phosphorylated sites of AGA: the structural impact of C163S mutation in aspartylglucosaminuria through molecular dynamics simulation.
Validation of Aspartylglucosaminidase Activity Assay for Human Serum Samples: Establishment of a Biomarker for Diagnostics and Clinical Studies.
📚 EuropePMC120 artigos no totalmostrando 39
Congenital Dermal Melanocytosis Associated With Aspartylglucosaminuria: Expanding the Dermatological Phenotype of a Rare Oligosaccharidosis.
The Australasian journal of dermatologyA 37-Year-Old Man With Intellectual Disability Discovered to Have Aspartylglucosaminuria: Implications for the Diagnosis of Genetic Causes.
Neurology. GeneticsGenetically Confirmed Case of Aspartylglycosaminuria (AGU).
Indian journal of pediatricsA new horizon in the phosphorylated sites of AGA: the structural impact of C163S mutation in aspartylglucosaminuria through molecular dynamics simulation.
Journal of biomolecular structure & dynamicsValidation of Aspartylglucosaminidase Activity Assay for Human Serum Samples: Establishment of a Biomarker for Diagnostics and Clinical Studies.
International journal of molecular sciencesAnalysis of urinary oligosaccharide excretion patterns by UHPLC/HRAM mass spectrometry for screening of lysosomal storage disorders.
Journal of inherited metabolic disease[Analysis of genetic variant in a child with Aspartylglucosaminuria].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsTopological Structural Brain Connectivity Alterations in Aspartylglucosaminuria: A Case-Control Study.
AJNR. American journal of neuroradiologyDetection of Aspartylglucosaminuria Patients from Magnetic Resonance Images by a Machine-Learning-Based Approach.
Brain sciencesA cross-sectional natural history study of aspartylglucosaminuria.
JIMD reportsTowards Splicing Therapy for Lysosomal Storage Disorders: Methylxanthines and Luteolin Ameliorate Splicing Defects in Aspartylglucosaminuria and Classic Late Infantile Neuronal Ceroid Lipofuscinosis.
CellsCarlos II of Spain, 'The Bewitched': cursed by aspartylglucosaminuria?
BMJ neurology openEffectiveness of early hematopoietic stem cell transplantation in preventing neurocognitive decline in aspartylglucosaminuria: A case series.
JIMD reportsKnockout of the CMP-Sialic Acid Transporter SLC35A1 in Human Cell Lines Increases Transduction Efficiency of Adeno-Associated Virus 9: Implications for Gene Therapy Potency Assays.
CellsAspartylglucosaminuria: Clinical Presentation and Potential Therapies.
Journal of child neurologyA new UHPLC-MS/MS method for the screening of urinary oligosaccharides expands the detection of storage disorders.
Orphanet journal of rare diseasesPre-clinical Gene Therapy with AAV9/AGA in Aspartylglucosaminuria Mice Provides Evidence for Clinical Translation.
Molecular therapy : the journal of the American Society of Gene TherapyStatistical Permutation Test Reveals Progressive and Region-Specific Iron Accumulation in the Thalami of Children with Aspartylglucosaminuria.
Brain sciencesThe Role of Hematopoietic Cell Transplant in the Glycoprotein Diseases.
CellsSusceptibility-Weighted Imaging Findings in Aspartylglucosaminuria.
AJNR. American journal of neuroradiologyDetailed profile of cognitive dysfunction in children with aspartylglucosaminuria.
Journal of inherited metabolic diseaseThe T99K variant of glycosylasparaginase shows a new structural mechanism of the genetic disease aspartylglucosaminuria.
Protein science : a publication of the Protein SocietyOptical coherence tomography features in brothers with aspartylglucosaminuria.
Annals of clinical and translational neurologyUPLC-MS/MS Analysis of Urinary Free Oligosaccharides for Lysosomal Storage Diseases: Diagnosis and Potential Treatment Monitoring.
Clinical chemistryBiochemical and structural insights into an allelic variant causing the lysosomal storage disorder - aspartylglucosaminuria.
FEBS lettersUrine oligosaccharide screening by MALDI-TOF for the identification of NGLY1 deficiency.
Molecular genetics and metabolismAmlexanox provides a potential therapy for nonsense mutations in the lysosomal storage disorder Aspartylglucosaminuria.
Biochimica et biophysica acta. Molecular basis of diseaseBiochemical characterization and comparison of aspartylglucosaminidases secreted in venom of the parasitoid wasps Asobara tabida and Leptopilina heterotoma.
PloS oneCrystal structure of a mutant glycosylasparaginase shedding light on aspartylglycosaminuria-causing mechanism as well as on hydrolysis of non-chitobiose substrate.
Molecular genetics and metabolismDevelopment of a new tandem mass spectrometry method for urine and amniotic fluid screening of oligosaccharidoses.
Rapid communications in mass spectrometry : RCMFunctional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production.
International journal of molecular sciencesWhite Matter Microstructure and Subcortical Gray Matter Structure Volumes in Aspartylglucosaminuria; a 5-Year Follow-up Brain MRI Study of an Adolescent with Aspartylglucosaminuria and His Healthy Twin Brother.
JIMD reportsAspartylglucosaminuria caused by a novel homozygous mutation in the AGA gene was identified by an exome-first approach in a patient from Japan.
Brain & developmentAspartylglycosaminuria: a review.
Orphanet journal of rare diseasesIdentification of Small Molecule Compounds for Pharmacological Chaperone Therapy of Aspartylglucosaminuria.
Scientific reportsBrain MRI findings in two Turkish pediatric patients with aspartylglucosaminuria.
The neuroradiology journalMeasurement of Elevated Concentrations of Urine Keratan Sulfate by UPLC-MSMS in Lysosomal Storage Disorders (LSDs): Comparison of Urine Keratan Sulfate Levels in MPS IVA Versus Other LSDs.
JIMD reportsA NOVEL ASPARTYLGLUCOSAMINURIA MUTATION IN A PATIENT WITH CO-EXISTENCE OF GAUCHER DISEASE.
Genetic counseling (Geneva, Switzerland)Brain MRI findings in aspartylglucosaminuria.
Journal of neuroradiology = Journal de neuroradiologieAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Aspartilglicosaminúria.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Aspartilglicosaminúria
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Congenital Dermal Melanocytosis Associated With Aspartylglucosaminuria: Expanding the Dermatological Phenotype of a Rare Oligosaccharidosis.
- A 37-Year-Old Man With Intellectual Disability Discovered to Have Aspartylglucosaminuria: Implications for the Diagnosis of Genetic Causes.
- Genetically Confirmed Case of Aspartylglycosaminuria (AGU).
- A new horizon in the phosphorylated sites of AGA: the structural impact of C163S mutation in aspartylglucosaminuria through molecular dynamics simulation.
- Validation of Aspartylglucosaminidase Activity Assay for Human Serum Samples: Establishment of a Biomarker for Diagnostics and Clinical Studies.
- Aspartylglucosaminuria.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:93(Orphanet)
- OMIM OMIM:208400(OMIM)
- MONDO:0008830(MONDO)
- GARD:5854(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q4412533(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
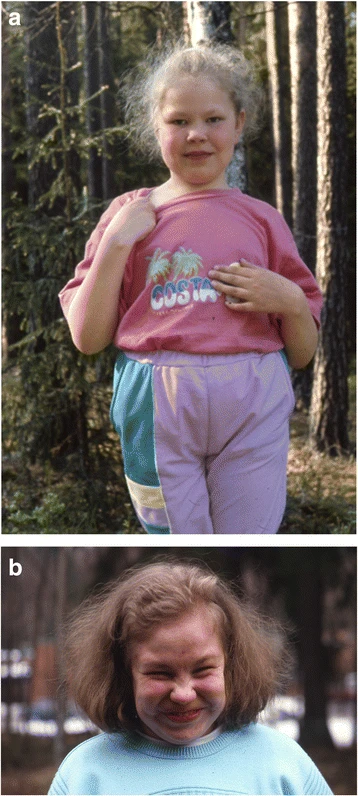
Aspartilglicosaminúria
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov