A condrodisplasia rizomélica é um tipo de condrodisplasia puntata. Esse é um grupo de doenças que tem como característica comum a presença de calcificações (pontinhos de cálcio) perto das juntas do bebê já ao nascer.
Introdução
O que você precisa saber de cara
A condrodisplasia rizomélica é um tipo de condrodisplasia puntata. Esse é um grupo de doenças que tem como característica comum a presença de calcificações (pontinhos de cálcio) perto das juntas do bebê já ao nascer.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 26 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 93 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
4 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Receptor that mediates peroxisomal import of proteins containing a C-terminal PTS1-type tripeptide peroxisomal targeting signal (SKL-type) (PubMed:11101887, PubMed:11336669, PubMed:12456682, PubMed:16314507, PubMed:17157249, PubMed:17428317, PubMed:21976670, PubMed:26344566, PubMed:7706321, PubMed:7719337, PubMed:7790377). Binds to cargo proteins containing a PTS1 peroxisomal targeting signal in the cytosol, and translocates them into the peroxisome matrix by passing through the PEX13-PEX14 dock
Cytoplasm, cytosolPeroxisome matrix
Peroxisome biogenesis disorder 2A
A fatal peroxisome biogenesis disorder belonging to the Zellweger disease spectrum and characterized clinically by severe neurologic dysfunction with profound psychomotor retardation, severe hypotonia and neonatal seizures, craniofacial abnormalities, liver dysfunction, and biochemically by the absence of peroxisomes. Additional features include cardiovascular and skeletal defects, renal cysts, ocular abnormalities, and hearing impairment. Most severely affected individuals with the classic form of the disease (classic Zellweger syndrome) die within the first year of life.
Dihydroxyacetonephosphate acyltransferase catalyzing the first step in the biosynthesis of plasmalogens, a subset of phospholipids that differ from other glycerolipids by having an alkyl chain attached through a vinyl ether linkage at the sn-1 position of the glycerol backbone, and which unique physical properties have an impact on various aspects of cell signaling and membrane biology
Peroxisome membrane
Rhizomelic chondrodysplasia punctata 2
A form of rhizomelic chondrodysplasia punctata, a disease characterized by severely disturbed endochondral bone formation, rhizomelic shortening of femur and humerus, vertebral disorders, dwarfism, cataract, cutaneous lesions, facial dysmorphism, and severe intellectual disability with spasticity.
Receptor required for the peroxisomal import of proteins containing a C-terminal PTS2-type peroxisomal targeting signal (PubMed:11931631, PubMed:22057399, PubMed:25538232, PubMed:9090381). Specifically binds to cargo proteins containing a PTS2 peroxisomal targeting signal in the cytosol (PubMed:11931631, PubMed:22057399, PubMed:25538232). Cargo protein-binding triggers interaction with PEX5 and formation of a ternary complex composed of PEX5 and PEX7 along with PTS2-containing cargo proteins, wh
Cytoplasm, cytosolPeroxisome matrix
Peroxisome biogenesis disorder complementation group 11
A peroxisomal disorder arising from a failure of protein import into the peroxisomal membrane or matrix. The peroxisome biogenesis disorders (PBD group) are genetically heterogeneous with at least 14 distinct genetic groups as concluded from complementation studies. Include disorders are: Zellweger syndrome (ZWS), neonatal adrenoleukodystrophy (NALD), infantile Refsum disease (IRD), and classical rhizomelic chondrodysplasia punctata (RCDP). ZWS, NALD and IRD are distinct from RCDP and constitute a clinical continuum of overlapping phenotypes known as the Zellweger spectrum (PBD-ZSS).
Catalyzes the exchange of the acyl chain in acyl-dihydroxyacetonephosphate (acyl-DHAP) for a long chain fatty alcohol, yielding the first ether linked intermediate, i.e. alkyl-dihydroxyacetonephosphate (alkyl-DHAP), in the pathway of ether lipid biosynthesis
Peroxisome membranePeroxisome
Rhizomelic chondrodysplasia punctata 3
A form of rhizomelic chondrodysplasia punctata, a disease characterized by severely disturbed endochondral bone formation, rhizomelic shortening of femur and humerus, vertebral disorders, dwarfism, cataract, cutaneous lesions, facial dysmorphism, and severe intellectual disability with spasticity.
Variantes genéticas (ClinVar)
449 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 566 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
6 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Condrodisplasia pontuada tipo rizomélico
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
2 pesquisas recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
4 ensaios clínicos encontrados, 2 ativos.
Publicações mais relevantes
Unraveling PEX6: insights into very-long-chain fatty acid levels and peroxisome biogenesis disorders in pediatric populations.
Peroxisome biogenesis disorders (PBDs) are a genetically heterogeneous group of metabolic diseases caused by impaired peroxisome assembly and function. PBDs exhibit striking clinical variability, ranging from lethal neonatal forms (e.g., Zellweger spectrum disorders) to milder childhood-onset presentations such as rhizomelic chondrodysplasia punctata. While elevated levels of very-long-chain fatty acids (VLCFAs) remain a key diagnostic feature, the existence of unusual cases with normal plasma VLCFA levels highlight the limitations of relying solely on this biochemical marker for diagnosis. Genetic variations in PEX6, an important peroxisome biogenesis factor, contribute significantly to this phenotypic diversity, with missense variants often associated with less severe disease compared to truncating mutations. Recent studies further implicate dysregulated pexophagy-a targeted autophagic degradation of peroxisomes-in the underlying disease mechanisms. This review underscores the necessity for a multifaceted diagnostic approach that, thorough clinical assessment, detailed biochemical evaluation, and advanced molecular genetic testing, seeks to improve diagnostic accuracy and patient care, particularly in pediatric populations. Advancements in identifying novel biomarkers and targeted therapies offer promise for tailored interventions, underscoring the importance of precision medicine in optimizing outcomes for pediatric PBD patients.
Development and evaluation of RhizoQOL, a quality-of-life caregiver-reported survey for rhizomelic chondrodysplasia punctata, a rare peroxisomal disorder.
Rhizomelic chondrodysplasia punctata (RCDP) is a rare genetic disorder characterized by symptoms such as respiratory dysfunction, seizures, orthopedic issues, and neurodevelopmental delay. Potential therapeutics for RCDP warrant the development of clinical outcome assessments to assess the efficacy of treatment and the well-being of patients. Our study aimed to develop a valid quality-of-life (QOL) caregiver-reported survey instrument, RhizoQOL, to be used as a supportive endpoint in RCDP clinical trials. Development of the RhizoQOL survey tool included three RCDP caregiver focus groups to elicit concepts to serve as potential domains in a QOL survey instrument for RCDP, pilot survey development and initial testing, cognitive interviewing of revised survey drafts to determine content validity, as well as a three-month longitudinal study for reliability and internal consistency of the survey instrument. Twenty-eight caregivers participated in the focus groups, reporting that concepts that could be appropriate domains of QOL in RCDP include psychosocial behavior, feeding symptoms, mobility symptoms, respiratory symptoms, seizures and related activity, and impact of treatment. Following pilot survey testing (n = 22) and stakeholder feedback, a revised pilot survey instrument was administered to five caregivers for cognitive interviewing. This resulted in a revised survey instrument with 31 question items, six domains, and a 1-5 Likert scale item response assessing frequency or severity of event in the question item. Longitudinal testing (n = 18) of the revised survey instrument found the average response score was 1.98 ± 0.97 for all question items, and a Cronbach's alpha value of 0.856, suggesting strong intra-survey question reliability. Using individual question item results from reliability testing, linear regression modeling, and testing for required magnitude of significant treatment effects, eight question items were removed from the survey instrument, resulting in a total of 23 question items within 6 discrete domains. The final RhizoQOL survey instrument, consisting of 23 questions, assesses the symptoms and experiences of RCDP patients as observed by caregivers and serves as a novel clinical outcome assessment for RCDP therapeutic clinical trials to assess the impacts of RCDP and support the overall effectiveness of treatments.
First-In-Human Safety, Tolerability, and Pharmacokinetics of PPI-1011, a Synthetic Plasmalogen Precursor.
PPI-1011 is a synthetic plasmalogen precursor designed to augment plasmalogen levels in patients with Rhizomelic chondrodysplasia punctata (RCDP), an ultra-rare genetic disorder caused by a plasmalogen deficiency that results in significant physical and mental delays. We report here a Phase I, randomized, double-blind, placebo-controlled study that evaluated the safety, tolerability, and pharmacokinetics (PK) of single (10-100 mg/kg) and multiple (75 and 100 mg/kg/day) ascending doses of PPI-1011 in healthy adults. All treatment-emergent adverse events (TEAEs) were mild, monitorable, and resolved without intervention, suggesting no significant safety concerns. The most common TEAEs were gastrointestinal in both the placebo and PPI-1011 groups, suggesting they were likely related to the oil-based nature of the formulation. PK analysis confirmed that both single (25, 50, 75 and 100 mg/kg) and multiple-dose (75 and 100 mg/kg, once daily) administration of PPI-1011 significantly increased serum levels of the target plasmalogen (PlsEtn 16:0/22:6). With a once-daily regimen, PPI-1011 administration resulted in a sustained increase of PlsEtn 16:0/22:6 serum concentrations in healthy participants over a duration of 14 days and beyond.
Metabolomic Profiling Reveals Brain Lipid Alterations in PEX7-Deficient Models of Rhizomelic Chondrodysplasia Punctata.
Rhizomelic chondrodysplasia punctata type 1 (RCDP1) is a peroxisomal disorder characterized by skeletal shortening, intellectual disability, seizures, cataracts, and reduced lifespans. RCDP1 is caused by biallelic loss-of-function variants in PEX7, which encodes a protein required for importing select enzymes into the peroxisome matrix, including those essential for ether lipid synthesis (e.g., plasmalogens) and the branched-chain fatty acid catabolism. Plasmalogen deficiency is a hallmark of RCDP1 and other peroxisomal disorders, including RCDP types 2-5 (RCDP2-5) and Zellweger spectrum disorders (ZSD). Here, we performed comprehensive metabolomic profiling of clinical samples from RCDP patients and Pex7-deficient mouse models. We identified profound neurometabolic disturbances in the cerebral cortex and cerebellum of Pex7-deficient mice involving multiple lipid classes, including phosphatidylethanolamines (PEs), phosphatidylcholines (PCs), acylcarnitines, and sphingomyelins. Notably, many of these neurometabolic alterations were absent in patient and Pex7-deficient mouse plasma, indicating that plasma-based profiling can underrepresent the extent of CNS lipid remodeling. Overall, these findings reveal novel insights into neurometabolic adaptations to plasmalogen deficiency and suggest the potential involvement of additional pathways that may contribute to neurological dysfunction in RCDP.
A diagnostic and management odyssey of a rare case of rhizomelic chondrodysplasia punctata.
Rhizomelic chondrodysplasia punctata (RCDP) is one of the rare inherited peroxisomal disorders that belongs to the heterogeneous group of chondrodysplasias presenting with proximal shortening of the limbs and distinctive punctate calcifications of bones and cartilages. This disorder occurs due to mutations in the genes responsible for peroxisomal biosynthesis inherited commonly as autosomal recessive, though a few are reported as x-linked dominant and recessive. The diagnosis can be made on clinical findings along with a skeletal survey; however, sonography and genetic analysis are crucial for a definitive prenatal diagnosis in cases where RCDP is inherited within a family as there is no definitive cure for RCDP and the treatment revolves around the management of its manifestations and complications. We hereby report the case of a neonate with features of RCDP. Significant respiratory involvement in RCDP babies is linked with high mortality which was also observed in this case. PEX7-related rhizomelic chondrodysplasia punctata (PEX7-RCDP), a peroxisome biogenesis disorder (PBD), has a classic (severe) form and a nonclassic (mild) form. Classic (severe) PEX7-RCDP is characterized by proximal shortening of the humerus (rhizomelia) and to a lesser degree the femur, punctate calcifications in cartilage with epiphyseal and metaphyseal abnormalities (chondrodysplasia punctata, or CDP), coronal clefts of the vertebral bodies, and cataracts that are usually present at birth or appear in the first few months of life. Birth weight, length, and head circumference are often at the lower range of normal; postnatal growth deficiency is profound. Intellectual disability is severe, and most children develop seizures. Most affected children do not survive the first decade of life; a proportion die in the neonatal period. Nonclassic (mild) PEX7-RCDP is characterized by congenital or childhood cataracts, CDP or, infrequently, chondrodysplasia manifesting only as mild epiphyseal changes, joint contractures, neurobehavioral abnormalities, and milder intellectual disability and growth restriction than classic PEX7-RCDP. The diagnosis of PEX7-RCDP is established in a proband with suggestive clinical, radiographic, and laboratory findings and biallelic pathogenic variants in PEX7 identified on molecular genetic testing. Treatment of manifestations: In those with classic (severe) PEX7-RCDP, management is supportive and limited by the severe health issues present at birth and poor outcome. Dietary restriction of phytanic acid to avoid the consequences of phytanic acid accumulation over time may benefit individuals with mild PEX7-RCDP. Physical therapy to improve contractures; orthopedic procedures may improve function in some individuals; consider need for positioning and mobility devices; spinal decompression with or without fusion depending on region of compression and myelopathic signs; cataract extraction may restore and/or preserve vision; community vision services through early intervention or school district; poor feeding and recurrent aspiration may necessitate placement of a gastrostomy tube; developmental and educational support; standard treatment of seizures by an experienced neurologist; good pulmonary clearance and attention to respiratory function; influenza vaccine, RSV monoclonal antibody, and other interventions to prevent lung disease; management of congenital heart disease per cardiologist; dermatologist management of skin manifestations; management of kidney and urinary tract anomalies per nephrologist and/or urologist; social work and care coordination as needed. Surveillance: Annual measurement of phytanic acid levels in children with non-classic (mild) PEX7-RCDP; musculoskeletal assessment, assessment for kyphosis and scoliosis, and neurologic exam with assessment for seizures at each visit; vision assessment every three months until cataracts are detected and then as recommended by ophthalmologist; assessment of growth, nutritional status, safety of oral intake, development, behavior, and respiratory function at each visit; cardiac exam, echocardiogram, and EKG with frequency per treating cardiologist; urine and imaging studies to assess for kidney stones as needed; assessment for skin issues and family needs at each visit. PEX7-RCDP is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for a PEX7 pathogenic variant, each sib of an affected individual has at conception a 25% chance of inheriting both pathogenic variants and being affected, a 50% chance of inheriting one pathogenic variant and being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the PEX7 pathogenic variants have been identified in an affected family member, molecular genetic carrier testing of at-risk relatives and prenatal/preimplantation genetic testing are possible.
Publicações recentes
Unraveling PEX6: insights into very-long-chain fatty acid levels and peroxisome biogenesis disorders in pediatric populations.
Metabolomic Profiling Reveals Brain Lipid Alterations in PEX7-Deficient Models of Rhizomelic Chondrodysplasia Punctata.
A diagnostic and management odyssey of a rare case of rhizomelic chondrodysplasia punctata.
PEX7-Related Rhizomelic Chondrodysplasia Punctata.
A bovine model of rhizomelic chondrodysplasia punctata caused by a deep intronic splicing variant in the GNPAT gene.
📚 EuropePMC112 artigos no totalmostrando 61
Unraveling PEX6: insights into very-long-chain fatty acid levels and peroxisome biogenesis disorders in pediatric populations.
Annals of pediatric endocrinology & metabolismMetabolomic Profiling Reveals Brain Lipid Alterations in PEX7-Deficient Models of Rhizomelic Chondrodysplasia Punctata.
BiomoleculesA diagnostic and management odyssey of a rare case of rhizomelic chondrodysplasia punctata.
JPMA. The Journal of the Pakistan Medical AssociationA bovine model of rhizomelic chondrodysplasia punctata caused by a deep intronic splicing variant in the GNPAT gene.
Genetics, selection, evolution : GSEDevelopment and evaluation of RhizoQOL, a quality-of-life caregiver-reported survey for rhizomelic chondrodysplasia punctata, a rare peroxisomal disorder.
Orphanet journal of rare diseasesFirst-In-Human Safety, Tolerability, and Pharmacokinetics of PPI-1011, a Synthetic Plasmalogen Precursor.
Clinical and translational scienceBiallelic Deletion of PEX26 Exon 4 in a Boy with Phenotypic Features of both Zellweger Syndrome and Infantile Refsum Disease.
Molecular syndromologyHuman Genetics of Atrial Septal Defect.
Advances in experimental medicine and biologyPeroxisomal Localization of a Truncated HMG-CoA Reductase under Low Cholesterol Conditions.
BiomoleculesA new test method for biochemical analysis of plasmalogens in dried blood spots and erythrocytes from patients with peroxisomal disorders.
Journal of inherited metabolic diseaseNeonatal rhizomelic chondrodysplasia punctata type 2 caused by a novel homozygous variant in the GNPAT gene.
Clinical case reportsDifferential Eye Expression of Xenopus Acyltransferase Gnpat and Its Biochemical Characterization Shed Light on Lipid-Associated Ocular Pathologies.
Investigative ophthalmology & visual scienceQuantitative analysis of ethanolamine plasmalogen species in red blood cells using liquid chromatography tandem mass spectrometry for diagnosing peroxisome biogenesis disorders.
Clinica chimica acta; international journal of clinical chemistryOverlapping and Distinct Features of Cardiac Pathology in Inherited Human and Murine Ether Lipid Deficiency.
International journal of molecular sciencesA Case of Rhizomelic Chondrodysplasia Punctata in a Neonate.
CureusAntenatal ultrasonographic diagnosis of rhizomelic chondrodysplasia punctata.
Journal of ultrasoundVery-Long-Chain Fatty Acids Quantification by Gas-Chromatography Mass Spectrometry.
Methods in molecular biology (Clifton, N.J.)Expanding the genotypic and phenotypic landscapes of rhizomelic chondrodysplasia punctata type 3 (RCDP3) with two novel families, and a review of the literature.
American journal of medical genetics. Part AA Pex7 Deficient Mouse Series Correlates Biochemical and Neurobehavioral Markers to Genotype Severity-Implications for the Disease Spectrum of Rhizomelic Chondrodysplasia Punctata Type 1.
Frontiers in cell and developmental biologyRhizomelic Chondro-Dysplasia Punctate (RCDP).
Indian pediatricsTricky Isomers-The Evolution of Analytical Strategies to Characterize Plasmalogens and Plasmanyl Ether Lipids.
Frontiers in cell and developmental biologyClinical diagnosis of metabolic disorders using untargeted metabolomic profiling and disease-specific networks learned from profiling data.
Scientific reportsA Novel Variant in the AGPS Gene Causes the Rare Rhizomelic Chondrodysplasia Punctata Type 3: A Case Report.
CureusPlasmalogens regulate the AKT-ULK1 signaling pathway to control the position of the axon initial segment.
Progress in neurobiologyGenetic epidemiology approach to estimating birth incidence and current disease prevalence for rhizomelic chondrodysplasia punctata.
Orphanet journal of rare diseasesA new GNPAT variant of foetal rhizomelic chondrodysplasia punctata.
Molecular genetics & genomic medicineTwins with PEX7 related intellectual disability and cataract: Highlighting phenotypes of peroxisome biogenesis disorder 9B.
American journal of medical genetics. Part AClinical, biochemical, and molecular characterization of mild (nonclassic) rhizomelic chondrodysplasia punctata.
Journal of inherited metabolic diseaseGeneral Movements and Developmental Functioning in an Individual with Rhizomelic Chondrodysplasia Punctata within the First Months of the Life: A Case Report.
Physical & occupational therapy in pediatricsConradi-Hünermann-Happle syndrome: report of a novel heterozygous mutation on the emopamil-binding protein gene, c.333delC.
Dermatology online journalGenome sequencing identifies a rare case of moderate Zellweger spectrum disorder caused by a PEX3 defect: Case report and literature review.
Molecular genetics and metabolism reportsChondrodysplasia punctata and neonatal lupus in an infant with positive anti-RNP and negative anti-Ro/SSA and -La/SSB antibodies, a case report.
Pediatric dermatologyUnequivocal Mapping of Molecular Ether Lipid Species by LC-MS/MS in Plasmalogen-Deficient Mice.
Analytical chemistryOral batyl alcohol supplementation rescues decreased cardiac conduction in ether phospholipid-deficient mice.
Journal of inherited metabolic diseaseOral administration of a synthetic vinyl-ether plasmalogen normalizes open field activity in a mouse model of rhizomelic chondrodysplasia punctata.
Disease models & mechanismsRhizomelic chondrodysplasia punctata: Role of EEG as a biomarker of impending epilepsy.
eNeurologicalSciRhizomelic chondrodysplasia punctata morbidity and mortality, an update.
American journal of medical genetics. Part AThe type-2 peroxisomal targeting signal.
Biochimica et biophysica acta. Molecular cell researchCervical Spine Deformities in Children With Rhizomelic Chondrodysplasia Punctata.
Journal of pediatric orthopedicsLeukodystrophy caused by plasmalogen deficiency rescued by glyceryl 1-myristyl ether treatment.
Brain pathology (Zurich, Switzerland)Application of machine learning algorithms for the differential diagnosis of peroxisomal disorders.
Journal of biochemistryStippled Calcifications over Bilateral Epiphyses of Humeri.
The Journal of pediatricsNeonatal Rhizomelic Chondrodysplasia Punctata Type 1: Weaving Evidence Into Clinical Practice.
The Journal of perinatal & neonatal nursing[Peroxisomal disorder, rhizomelyc chondrodysplasia punctata type 1: case report].
Revista chilena de pediatriaType 1 rhizomelic chondrodysplasia punctata with a homozygous PEX7 mutation.
Journal of pediatric endocrinology & metabolism : JPEMDrosophila Courtship Conditioning As a Measure of Learning and Memory.
Journal of visualized experiments : JoVEIdentification and diagnostic value of phytanoyl- and pristanoyl-carnitine in plasma from patients with peroxisomal disorders.
Molecular genetics and metabolismReduced muscle strength in ether lipid-deficient mice is accompanied by altered development and function of the neuromuscular junction.
Journal of neurochemistryRecombinant human dihydroxyacetonephosphate acyl-transferase characterization as an integral monotopic membrane protein.
Biochemical and biophysical research communicationsGrowth charts for individuals with rhizomelic chondrodysplasia punctata.
American journal of medical genetics. Part ARare Case of Rhizomelic Chondrodysplasia Punctata.
Journal of orthopaedic case reportsClinical and Biochemical Pitfalls in the Diagnosis of Peroxisomal Disorders.
NeuropediatricsPRENATAL DIAGNOSIS OF RHIZOMELIC CHONDRODYSPLASIA PUNCTATA.
Genetic counseling (Geneva, Switzerland)Peroxisomes in brain development and function.
Biochimica et biophysica actaWhole Exome Sequencing Reveals Compound Heterozygosity for Ethnically Distinct PEX7 Mutations Responsible for Rhizomelic Chondrodysplasia Punctata, Type 1.
Case reports in geneticsCongenital heart defects common in rhizomelic chondrodysplasia punctata.
American journal of medical genetics. Part APhytol in a pharma-medico-stance.
Chemico-biological interactionsA novel type of rhizomelic chondrodysplasia punctata, RCDP5, is caused by loss of the PEX5 long isoform.
Human molecular geneticsRhizomelic Chondrodysplasia Punctata Type 1 Caused by a Novel Mutation in the PEX7 Gene.
Journal of clinical research in pediatric endocrinologyRadiographic features of the skeleton in disorders of post-squalene cholesterol biosynthesis.
Pediatric radiologyTargeted carrier screening for four recessive disorders: high detection rate within a founder population.
European journal of medical geneticsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Condrodisplasia pontuada tipo rizomélico.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Condrodisplasia pontuada tipo rizomélico
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Unraveling PEX6: insights into very-long-chain fatty acid levels and peroxisome biogenesis disorders in pediatric populations.
- Development and evaluation of RhizoQOL, a quality-of-life caregiver-reported survey for rhizomelic chondrodysplasia punctata, a rare peroxisomal disorder.
- First-In-Human Safety, Tolerability, and Pharmacokinetics of PPI-1011, a Synthetic Plasmalogen Precursor.
- Metabolomic Profiling Reveals Brain Lipid Alterations in PEX7-Deficient Models of Rhizomelic Chondrodysplasia Punctata.
- A diagnostic and management odyssey of a rare case of rhizomelic chondrodysplasia punctata.
- PEX7-Related Rhizomelic Chondrodysplasia Punctata.
- A bovine model of rhizomelic chondrodysplasia punctata caused by a deep intronic splicing variant in the GNPAT gene.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:177(Orphanet)
- MONDO:0015776(MONDO)
- GARD:13160(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q7320761(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
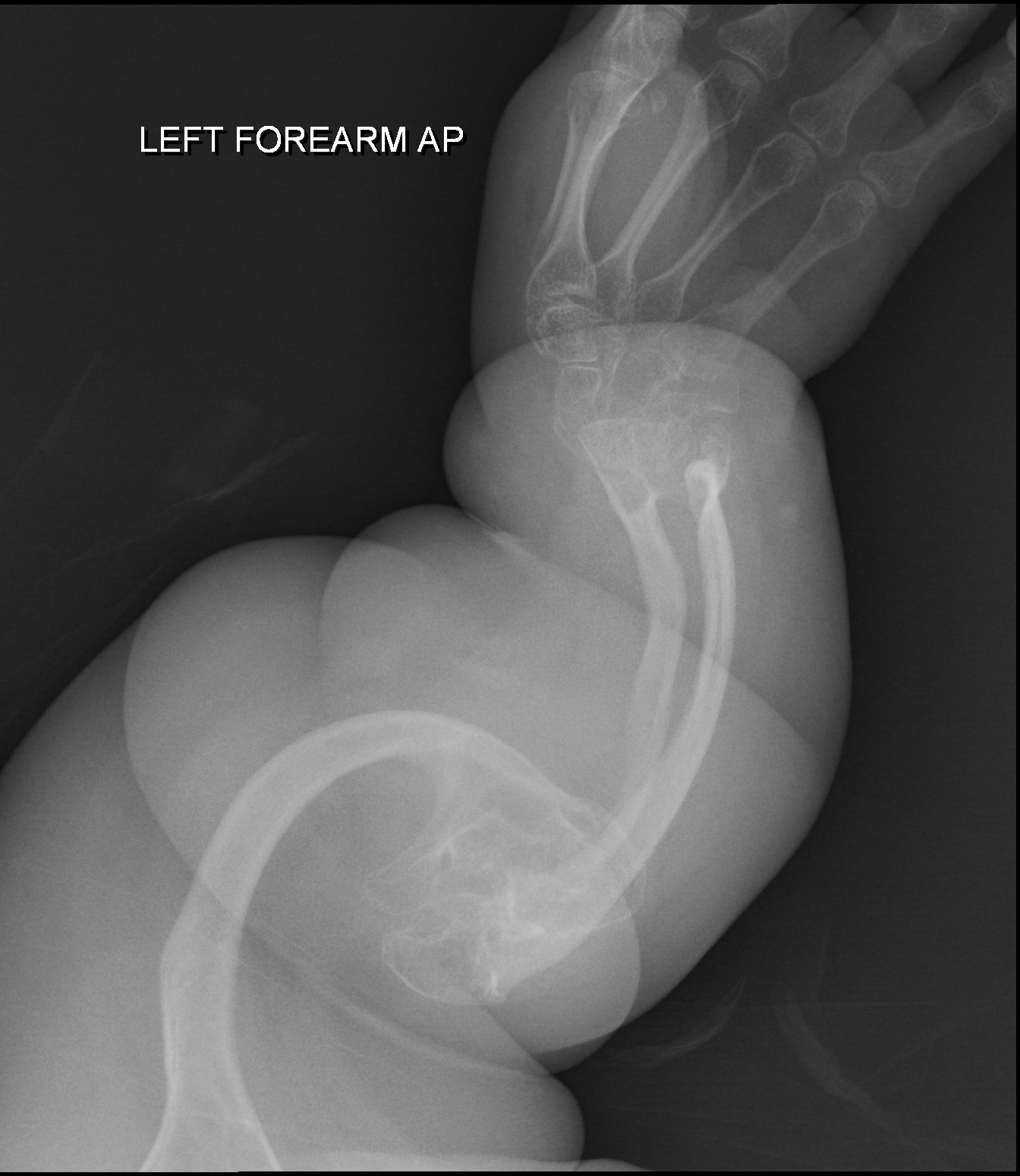
Condrodisplasia pontuada tipo rizomélico
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov