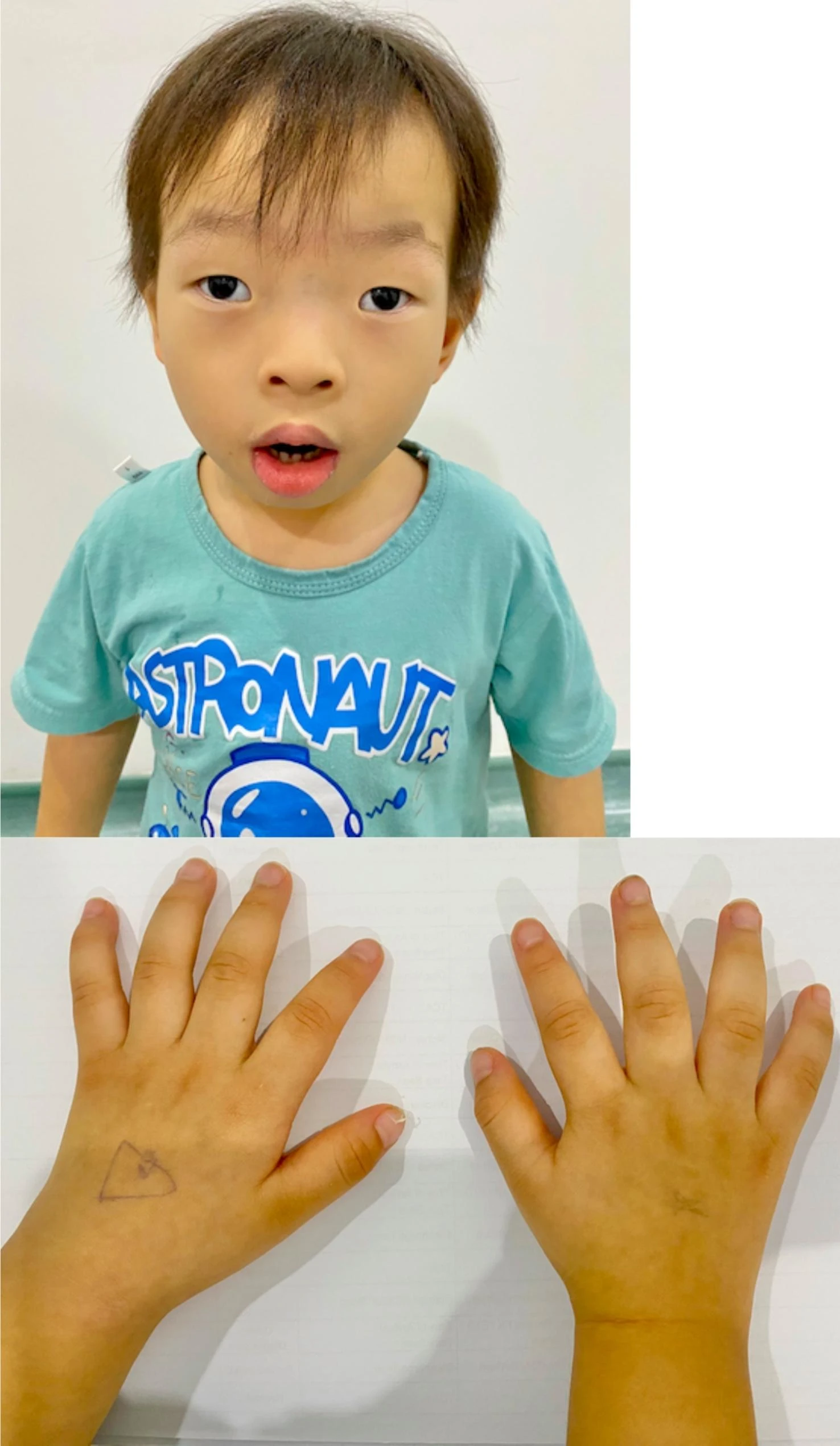
É uma deficiência intelectual rara, ligada ao cromossomo X, que faz parte de uma síndrome. Ela é caracterizada por atraso no desenvolvimento geral, crescimento lento após o nascimento que leva a baixa estatura, características faciais distintas, mãos pequenas com dedos que afinam na ponta, e problemas progressivos nos ossos, incluindo curvatura anormal da coluna (cifoescoliose) e deformidades no tórax (como "peito de pombo" ou "peito escavado"). A deficiência intelectual pode variar de leve a grave.
Introdução
O que você precisa saber de cara
É uma deficiência intelectual rara, ligada ao cromossomo X, que faz parte de uma síndrome. Ela é caracterizada por atraso no desenvolvimento geral, crescimento lento após o nascimento que leva a baixa estatura, características faciais distintas, mãos pequenas com dedos que afinam na ponta, e problemas progressivos nos ossos, incluindo curvatura anormal da coluna (cifoescoliose) e deformidades no tórax (como "peito de pombo" ou "peito escavado"). A deficiência intelectual pode variar de leve a grave.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 31 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 113 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: X-linked dominant.
Curadoria gene-doença
fontes oficiaisSerine/threonine-protein kinase that acts downstream of ERK (MAPK1/ERK2 and MAPK3/ERK1) signaling and mediates mitogenic and stress-induced activation of the transcription factors CREB1, ETV1/ER81 and NR4A1/NUR77, regulates translation through RPS6 and EIF4B phosphorylation, and mediates cellular proliferation, survival, and differentiation by modulating mTOR signaling and repressing pro-apoptotic function of BAD and DAPK1 (PubMed:16213824, PubMed:16223362, PubMed:17360704, PubMed:9770464). In f
NucleusCytoplasm
Coffin-Lowry syndrome
An X-linked disorder characterized by intellectual disability associated with facial and digital dysmorphisms, progressive skeletal malformations, growth retardation, hearing deficit and paroxysmal movement disorders.
Variantes genéticas (ClinVar)
624 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 321 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
7 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Coffin-Lowry
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Case report of breastfeeding after maternal iodine contrast: neonatal hypothyroidism revealing an underlying congenital disorder.
Iodine plays a critical role in producing thyroid hormones essential for brain development. An imbalance of iodine, whether deficiency or excess, can disrupt thyroid function. Iodine-induced hypothyroidism is rare but has been reported, particularly in premature infants exposed to excess iodine. Currently, iohexol, a nonionic radiocontrast agent, has limited data but is considered compatible with breastfeeding. A small for gestation African American male neonate was born at 36 weeks and 3 days of gestation and admitted to the neonatal intensive care unit for respiratory distress and prematurity in the United States. Samples for Illinois newborn screens done at admission and repeated after 48 h of life were negative for thyroid abnormalities. On the infant's fifth day of life, the mother underwent a contrast-enhanced imaging study using iohexol, after which the infant received breast milk from days 5-11. On day 11, the neonate had an elevated thyroid-stimulating hormone (TSH) and low free thyroxine (T4) levels, consistent with hypothyroidism. Urine iodine level in the infant was checked and found to be elevated, prompting concern for the exposure to iodine in breastmilk. However, the need to increase the levothyroxine dose to achieve normal thyroid levels was not consistent with a transient effect such as iodine exposure. Genetic testing revealed a likely pathogenic intragenic deletion in the gene RPS6KA3, consistent with Coffin-Lowry syndrome, as well as two variants of unknown significance (VUS) in IYD. This case highlights the complex interplay between genetic factors and environmental influences in the development of neonatal hypothyroidism. While iodine contrast exposure through breast milk is generally considered safe, this case underscores the potential risks in preterm infants. Initially, iodine exposure through breastmilk was considered a likely contributor to thyroid dysfunction, but with further time and evaluation, congenital thyroid dysgenesis with underlying genetic findings complicated the diagnosis. This case demonstrates the importance of a comprehensive evaluation when working up thyroid dysfunction to exclude other potential etiologies before interrupting breastfeeding.
A 2-year-old girl with merged phenotypes: galactosemia and Coffin-Lowry syndrome.
Galactosemia is a congenital disorder of carbohydrate metabolism, in which the body is unable to metabolize galactose properly. Coffin-Lowry syndrome (CLS) is characterized by intellectual disability, developmental delay, dysmorphic features, growth retardation, vision and hearing loss, and skeletal changes, which is an X-linked disorder, with males being more severely affected, whereas the clinical findings in females show variability. This case is presented due to the rare concomitance of galactosemia and CLS. A 2-year-old female patient, previously diagnosed with galactosemia, who had good dietary adherence was noticed to have developmental delay, dysmorphic features, nephrolithiasis and recurrent pericardial effusions during follow-up. Further research was carried out to diagnose an underlying second disease. Metabolic tests were inconclusive. Clinical exome sequencing (CES) analysis, revealed a heterozygous c.472C>T p. (Arg158Cys) pathogenic variant in RPS6KA3 (OMIM #300075) and CLS (OMIM #303600) was diagnosed. This case report is a unique summary of a patient with galactosemia who further was diagnosed with CLS that emphasizes the possibility of co-occurrence of rare diseases and highlights the importance of conducting further investigations in patients with unexplained findings in the context of existing metabolic diseases.
[Case Report of One Family With Coffin-Lowry Syndrome and Literature Review of 28 Cases in China].
To investigate the clinical phenotypes and genotypic characteristics of Chinese patients with Coffin-Lowry syndrome. The clinical data and genetic test results of a family with Coffin-Lowry syndrome were retrospectively analyzed. A literature review was conducted to summarize the clinical characteristics and gene mutation characteristics of patients with Coffin-Lowry syndrome in China. The proband was a 1-year-old boy with distinctive facial features, puffy but tapered fingers, hypotonia, growth retardation, and delayed cognitive and motor development. Genetic analysis revealed a hemizygous c.1603-2A>G mutation in intron 17 of the RPS6KA3 gene in the proband. His mother was a heterozygous carrier. The identified mutation has not been reported previously. The proband's maternal half-brother and half-sister also exhibited similar clinical manifestations and were diagnosed with Coffin-Lowry syndrome together with the proband. The proband was followed up until 3 years and 8 months old, by which time he was not capable of walking steadily independently or speech. Including the 4 members of this family, a total of 28 Chinese patients were identified. Their clinical manifestations included special facial features (100%), cognitive and language/motor developmental delays (92.6%), hypotonia (95.2%), tapered fingers (88.5%), and scoliosis or kyphosis (45%). Genetic sequencing was performed in 24 patients, revealing missense mutations in 3 cases (12.5%), frameshift mutations in 5 cases (20.8%), nonsense mutations in 9 cases (37.5%), splice-site mutations in 4 cases (16.7%), and exon deletions in 2 cases (8.3%). No mutation hotspots were identified. Coffin-Lowry syndrome should be considered in children with cognitive and language/motor developmental delays, distinctive facial features, tapered fingers, and hypotonia. Genetic testing can assist with early diagnosis. 探讨中国Coffin-Lowry 综合征患者的临床表型及基因型特征。 回顾一个Coffin-Lowry 综合征家系的临床资料及基因检测结果,并复习文献,总结中国Coffin-Lowry 综合征患者的临床特点及基因突变特点。 患儿,男,1岁,有特殊面容、锥形手指、肌张力低下、生长迟缓,认知运动发育落后。患儿RPS6KA3基因17号内含子上发生c.1603-2A>G半合子突变,母亲发生杂合突变,该变异是首次报告。其同母异父的哥哥和姐姐也有相似的临床表现,与患儿一并诊断为Coffin-Lowry综合征。该患儿随访到3岁8月,独走不稳,无语言出现。包括该家系4人,共有28例中国患者,表现为特殊面容(100%),认知及语言运动发育迟滞(92.6%)、肌张力低(95.2%)、锥形手指(88.5%)、脊柱侧弯/后凸(45%)等。共有24个患者行基因测序,其中错义突变3例( 12.5%),移码突变5例(20.8%),无义突变9例(37.5%),剪接突变4例(16.7%),外显子缺失2例(8.3%),未出现变异热点。 患儿有认知及语言运动发育迟缓,特殊面容伴有锥形手指、肌张力低下时应考虑Coffin-Lowry 综合征;基因检测有助于早期诊断。
Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder.
The ribosomal protein S6 kinase family members play essential biological functions in disease, from cancer to intellectual disability. Little is known about ribosomal proteins S6 kinase C1 (RPS6KC1), aside from its lack of phosphorylation capacity and its roles in sphingosine-1-phosphate signaling and peroxiredoxin-3 (PRDX3) transport to mitochondria. Through whole-exome sequencing, we identified bi-allelic RPS6KC1 variants in 13 individuals from 8 independent families. Phenotypic manifestations included neurodevelopmental delay, hypotonia, spastic paraplegia, brain white matter loss, and dysmorphic features overlapping with Coffin-Lowry syndrome caused by RPS6KA3 mutations. Functional studies on peripheral blood mononuclear cells (PBMCs) from the different individuals indicated diminished expression and phosphorylation of RPS6, impacting ribosomal protein synthesis, and a decrease in the known interactors PRDX3 and sphingosine kinase 1 (SPHK1), accompanied by marked repression of the mammalian target of rapamycin (mTOR)/phosphatidylinositol 3-kinase (PI3K) pathway. We detected a dysregulation of phosphoinositides and sphingoid base levels in plasma samples from the different individuals. Further studies in HAP1 RPS6KC1-knockdown cells suggested that RPS6KC1 may regulate PRDX3 and SPHK1 activities by facilitating their endosome anchoring. In Drosophila melanogaster, the knockdown of CG7156, the RPS6KC1 ortholog, resulted in locomotor dysfunction, defective neuromuscular junctions, reduced lifespan, and decreased mTOR activity. Overexpression of mTOR in this model improved motor function and lifespan. These findings underscore the crucial roles of RPS6KC1 in neurodevelopment by controlling ribosomal protein synthesis, lipid signaling, and the mTOR pathway.
Coffin-Lowry Syndrome: A Case of Clinical Convergence for Psychology, Neuropsychology, Psychiatry, Genetics, and Neurology.
We present the case of a 15-year-old girl with new-onset psychosis and abnormal white matter activity on neuroimaging, engaging multidisciplinary care between genetics, neurology, psychiatry, and neuropsychology. She functioned well in mainstream education despite below average intellectual functioning. Physical examination findings enabled the diagnosis, and patient improved with joint psychological and behavioral outpatient services.
Publicações recentes
Challenges in Diagnosis and Management of Coffin-Lowry Syndrome-Single-Center Experience.
Case report of breastfeeding after maternal iodine contrast: neonatal hypothyroidism revealing an underlying congenital disorder.
Coffin-Lowry syndrome: a systematic review of RPS6KA3 confirmed cases and implications for diagnosis and counseling.
[Case Report of One Family With Coffin-Lowry Syndrome and Literature Review of 28 Cases in China].
🥇 Revisão sistemáticaCardiovascular Collapse During Scoliosis Surgery in a Patient With Coffin-Lowry Syndrome and Mesocardia.
🥇 Ensaio randomizado📚 EuropePMC169 artigos no totalmostrando 72
Case report of breastfeeding after maternal iodine contrast: neonatal hypothyroidism revealing an underlying congenital disorder.
International breastfeeding journalCoffin-Lowry syndrome: a systematic review of RPS6KA3 confirmed cases and implications for diagnosis and counseling.
Frontiers in genetics[Case Report of One Family With Coffin-Lowry Syndrome and Literature Review of 28 Cases in China].
Sichuan da xue xue bao. Yi xue ban = Journal of Sichuan University. Medical science editionCardiovascular Collapse During Scoliosis Surgery in a Patient With Coffin-Lowry Syndrome and Mesocardia.
CureusBi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder.
American journal of human geneticsA 2-year-old girl with merged phenotypes: galactosemia and Coffin-Lowry syndrome.
Journal of pediatric endocrinology & metabolism : JPEMRSK2 and its binding partners: an emerging signaling node in cancers.
Archives of pharmacal researchMyoclonic reflex and non-reflex seizures in a female child with Coffin-Lowry syndrome: Clinical vignette.
Epileptic disorders : international epilepsy journal with videotapeCoffin-Lowry Syndrome: A Case of Clinical Convergence for Psychology, Neuropsychology, Psychiatry, Genetics, and Neurology.
Journal of child neurologyThe natural course of newborns with transient congenital hypothyroidism.
Endocrine connectionsAirway management of a patient with coffin-lowry syndrome: a case report.
BMC anesthesiologyChewing and swallowing training in Coffin-Lowry syndrome: A case report.
Journal of oral rehabilitationIdentification of RSK substrates using an analog-sensitive kinase approach.
The Journal of biological chemistryAn unexpected presentation of very severe hypertriglyceridemia in a boy with Coffin-Lowry syndrome: a case report.
BMC pediatricsEstablishment of linkage phase, using Oxford Nanopore Technologies, for preimplantation genetic testing of Coffin-Lowry syndrome with a de novo RPS6KA3 mutation.
Frontiers in geneticsDelayed postnatal brain development and ontogenesis of behavior and cognition in a mouse model of intellectual disability.
Neurobiology of diseaseExploring the impacts of a coffin-lying experience on life and death attitudes of medical and nursing students: preliminary findings.
BMC medical educationDefective synaptic plasticity in a model of Coffin-Lowry syndrome is rescued by simultaneously targeting PKA and MAPK pathways.
Learning & memory (Cold Spring Harbor, N.Y.)Novel RPS6KA3 mutations cause Coffin-Lowry syndrome in two patients and concurrent compulsive eyebrow-pulling behavior in one of them.
Psychiatric geneticsMitral valve repair and tricuspid annuloplasty for Coffin-Lowry syndrome.
Asian cardiovascular & thoracic annalsCase Report: Chinese female patients with a heterozygous pathogenic RPS6KA3 gene variant c.898C>T and distal 22q11.2 microdeletion.
Frontiers in geneticsCoffin-Lowry Syndrome Induced by RPS6KA3 Gene Variation in China: A Case Report in Twins.
Medicina (Kaunas, Lithuania)Short Bones, Renal Stones, and Diagnostic Moans: Hypercalcemia in a Girl Found to Have Coffin-Lowry Syndrome.
Journal of investigative medicine high impact case reportsPremature Loss of Deciduous Teeth as a Symptom of Systemic Disease: A Narrative Literature Review.
International journal of environmental research and public healthFirst female Korean child with Coffin-Lowry syndrome: a novel variant in RPS6KA3 diagnosed by exome sequencing and a literature review.
Annals of pediatric endocrinology & metabolismIdentification of a New Mutation in RSK2, the Gene for Coffin-Lowry Syndrome (CLS), in Two Related Patients with Mild and Atypical Phenotypes.
Brain sciencesQuantitative description of the interactions among kinase cascades underlying long-term plasticity of Aplysia sensory neurons.
Scientific reportsAccelerated tooth movement in Rsk2-deficient mice with impaired cementum formation.
International journal of oral scienceModeling suggests combined-drug treatments for disorders impairing synaptic plasticity via shared signaling pathways.
Journal of computational neuroscienceNovel missense mutation c.1784A>G, p.Tyr595Cys in RPS6KA3 gene responsible for Coffin-Lowry syndrome in a family with variable features and diabetes 2.
Clinical dysmorphology[Prenatal diagnosis and genetic analysis of a fetus with Xp22.12 microduplication].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCoffin-Lowry syndrome in a girl with 46,XX,t(X;11)(p22;p15)dn: Identification of RPS6KA3 disruption by whole genome sequencing.
Clinical case reportsCustomised next-generation sequencing multigene panel to screen a large cohort of individuals with chromatin-related disorder.
Journal of medical geneticsRole of p90 ribosomal S6 kinase in long-term synaptic facilitation and enhanced neuronal excitability.
Scientific reportsBRM: the core ATPase subunit of SWI/SNF chromatin-remodelling complex-a tumour suppressor or tumour-promoting factor?
Epigenetics & chromatinA new missense mutation in DPF2 gene related to Coffin Siris syndrome 7: Description of a mild phenotype expanding DPF2-related clinical spectrum and differential diagnosis among similar syndromes epigenetically determined.
Brain & developmentGenome-wide association analysis for lethal brachycephalic-like facial dysmorphia in Labrador Retrievers.
Animal geneticsAn unusual cause for Coffin-Lowry syndrome: Three brothers with a novel microduplication in RPS6KA3.
American journal of medical genetics. Part A[Analysis of RPS6KA3 gene mutation in a Chinese pedigree affected with Coffin-Lowry syndrome].
Zhonghua yi xue yi chuan xue za zhi = Zhonghua yixue yichuanxue zazhi = Chinese journal of medical geneticsCoffin-Lowry syndrome in Chinese.
American journal of medical genetics. Part AA Bizarre Gait as a Result of Overlapping Functional Disorder With Coffin-Lowry Syndrome.
Movement disorders clinical practiceSevere Restrictive Lung Disease in One of the Oldest Documented Males With Coffin-Lowry Syndrome.
Journal of investigative medicine high impact case reportsGrowth Concerns in Coffin-Lowry Syndrome: A Case Report and Literature Review.
Frontiers in pediatricsDe Novo SOX4 Variants Cause a Neurodevelopmental Disease Associated with Mild Dysmorphism.
American journal of human geneticsAnimal Models for Coffin-Lowry Syndrome: RSK2 and Nervous System Dysfunction.
Frontiers in behavioral neuroscienceDrosophila RSK Influences the Pace of the Circadian Clock by Negative Regulation of Protein Kinase Shaggy Activity.
Frontiers in molecular neurosciencePeriventricular small cystic lesions in a patient with Coffin-Lowry syndrome who exhibited a novel mutation in the RPS6KA3 gene.
Brain & developmentSelective alteration of adult hippocampal neurogenesis and impaired spatial pattern separation performance in the RSK2-deficient mouse model of Coffin-Lowry syndrome.
Neurobiology of diseaseThe natural history of spinal deformity in patients with Coffin-Lowry syndrome.
Journal of children's orthopaedicsPerioperative management of a patient with Coffin-Lowry syndrome complicated by severe obesity: A case report and literature review.
MedicineA Highlighted Case for Emphasizing on Clinical Diagnosis for Rare Syndrome in Third World.
Iranian journal of child neurologyMolecular Targeting of ERKs/RSK2 Signaling in Cancers.
Current pharmaceutical designPeculiar Clinical Presentation of Coxsackievirus B4 Infection: Neonatal Restrictive Cardiomyopathy.
AJP reportsRsk2 Knockout Affects Emotional Behavior in the IntelliCage.
Behavior geneticsHigh diagnostic yield of clinically unidentifiable syndromic growth disorders by targeted exome sequencing.
Clinical geneticsForamen magnum compression in Coffin-Lowry syndrome: A case report.
American journal of medical genetics. Part AMechanical ventilation in Coffin-Lowry syndrome: a case report.
Revista Brasileira de terapia intensivaExome sequencing in children of women with skewed X-inactivation identifies atypical cases and complex phenotypes.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology SocietyBacterial Expression, Purification and In Vitro Phosphorylation of Full-Length Ribosomal S6 Kinase 2 (RSK2).
PloS oneEight years of follow-up after laminectomy of calcium pyrophosphate crystal deposition in the cervical yellow ligament of patient with Coffin-Lowry syndrome: A case report.
MedicineRsk2, the Kinase Mutated in Coffin-Lowry Syndrome, Controls Cementum Formation.
Journal of dental researchDrop episodes improved after tracheotomy: a case of Coffin-Lowry syndrome associated with obstructive sleep apnea syndrome.
European review for medical and pharmacological sciencesDifficult airway management using the Pentax-AWS Airwayscope with a thin Intlock and bronchofiberscope in a patient with Coffin-Lowry syndrome.
Journal of clinical anesthesiaLoss of the Coffin-Lowry syndrome-associated gene RSK2 alters ERK activity, synaptic function and axonal transport in Drosophila motoneurons.
Disease models & mechanisms625 kb microduplication at Xp22.12 including RPS6KA3 in a child with mild intellectual disability.
Journal of human geneticsConcomitant partial exon skipping by a unique missense mutation of RPS6KA3 causes Coffin-Lowry syndrome.
GeneRecurrent Nonconvulsive Status Epilepticus in a Patient with Coffin-Lowry Syndrome.
Molecular syndromologyMyoclonus in childhood-onset neurogenetic disorders: The importance of early identification and treatment.
European journal of paediatric neurology : EJPN : official journal of the European Paediatric Neurology Society[Deletion of the RPS6KA3 gene in a female with a classical phenotype of Coffin-Lowry syndrome including stimulus-induced drop attacks].
Revista de neurologiaA familial case of Coffin-Lowry syndrome caused by RPS6KA3 C.898C>T mutation associated with multiple abnormal brain imaging findings.
Genetic counseling (Geneva, Switzerland)Consider the neuro-cardiac continuum of Coffin-Lowry syndrome!
American journal of medical genetics. Part AStimulus-induced myoclonus treated effectively with clonazepam in genetically confirmed Coffin-Lowry syndrome.
Epilepsy & behavior case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Coffin-Lowry.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Coffin-Lowry
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Case report of breastfeeding after maternal iodine contrast: neonatal hypothyroidism revealing an underlying congenital disorder.
- A 2-year-old girl with merged phenotypes: galactosemia and Coffin-Lowry syndrome.
- [Case Report of One Family With Coffin-Lowry Syndrome and Literature Review of 28 Cases in China].Sichuan da xue xue bao. Yi xue ban = Journal of Sichuan University. Medical science edition· 2025· PMID 41536659mais citado
- Bi-allelic variants in the ribosomal protein RPS6KC1 cause a complex neurodevelopmental disorder.
- Coffin-Lowry Syndrome: A Case of Clinical Convergence for Psychology, Neuropsychology, Psychiatry, Genetics, and Neurology.
- Challenges in Diagnosis and Management of Coffin-Lowry Syndrome-Single-Center Experience.
- Coffin-Lowry syndrome: a systematic review of RPS6KA3 confirmed cases and implications for diagnosis and counseling.
- Cardiovascular Collapse During Scoliosis Surgery in a Patient With Coffin-Lowry Syndrome and Mesocardia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:192(Orphanet)
- OMIM OMIM:303600(OMIM)
- MONDO:0010561(MONDO)
- GARD:6123(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q1106881(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Coffin-Lowry
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Ensaios clínicos
- fonte: ClinicalTrials.gov