Forma autossômica recessiva de osteopetrose causada por mutação(ões) em pelo menos 8 genes relacionados à função dos osteoclastos. Esta condição é caracterizada pela falha dos osteoclastos em reabsorver o osso, resultando em modelagem/remodelação óssea prejudicada e fragilidade esquelética, apesar do aumento da massa óssea; também está associado a insuficiência hematopoiética, hipocalcemia, erupção dentária perturbada, síndromes de compressão nervosa e comprometimento do crescimento. Alguns casos também estão associados à deterioração neurológica progressiva.
Introdução
O que você precisa saber de cara
Forma autossômica recessiva de osteopetrose causada por mutação(ões) em pelo menos 8 genes relacionados à função dos osteoclastos. Esta condição é caracterizada pela falha dos osteoclastos em reabsorver o osso, resultando em modelagem/remodelação óssea prejudicada e fragilidade esquelética, apesar do aumento da massa óssea; também está associado a insuficiência hematopoiética, hipocalcemia, erupção dentária perturbada, síndromes de compressão nervosa e comprometimento do crescimento. Alguns casos também estão associados à deterioração neurológica progressiva.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 69 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 202 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
10 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisSubunit of the V0 complex of vacuolar(H+)-ATPase (V-ATPase), a multisubunit enzyme composed of a peripheral complex (V1) that hydrolyzes ATP and a membrane integral complex (V0) that translocates protons (By similarity). V-ATPase is responsible for acidifying and maintaining the pH of intracellular compartments and in some cell types, is targeted to the plasma membrane, where it is responsible for acidifying the extracellular environment (By similarity). Seems to be directly involved in T-cell a
Membrane
Osteopetrosis, autosomal recessive 1
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Sodium-independent anion exchanger which mediates the electroneutral exchange of chloride for bicarbonate ions across the cell membrane (PubMed:15184086, PubMed:34668226). Plays an important role in osteoclast differentiation and function (PubMed:34668226). Regulates bone resorption and calpain-dependent actin cytoskeleton organization in osteoclasts via anion exchange-dependent control of pH (By similarity). Essential for intracellular pH regulation in CD8(+) T-cells upon CD3 stimulation, modul
Apical cell membraneBasolateral cell membrane
Osteopetrosis, autosomal recessive 9
A form of osteopetrosis, a rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB9 is characterized by increased bone density and bone fragility, as well as renal failure.
Acts as a multivalent adapter protein that regulates Rab7-dependent and HOPS complex-dependent fusion events in the endolysosomal system and couples autophagic and the endocytic trafficking pathways. Acts as a dual effector of RAB7A and ARL8B that simultaneously binds these GTPases, bringing about clustering and fusion of late endosomes and lysosomes (PubMed:25498145, PubMed:28325809). Required for late stages of endolysosomal maturation, facilitating both endocytosis-mediated degradation of gro
Autolysosome membraneEndosome membraneLate endosome membraneLysosome membrane
Osteopetrosis, autosomal recessive 6
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Receptor for TNFSF11/RANKL/TRANCE/OPGL; essential for RANKL-mediated osteoclastogenesis (PubMed:9878548). Its interaction with EEIG1 promotes osteoclastogenesis via facilitating the transcription of NFATC1 and activation of PLCG2 (By similarity). Involved in the regulation of interactions between T-cells and dendritic cells (By similarity)
Cell membraneMembrane raft
Familial expansile osteolysis
Rare autosomal dominant bone disorder characterized by focal areas of increased bone remodeling. The osteolytic lesions develop usually in the long bones during early adulthood. FEO is often associated with early-onset deafness and loss of dentition.
Required for osteoclast and melanocyte maturation and function
Lysosome membrane
Osteopetrosis, autosomal recessive 5
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB5 patients manifest primary central nervous system involvement in addition to the classical stigmata of severe bone sclerosis, growth failure, anemia, thrombocytopenia and visual impairment with optic atrophy.
Catalyzes the reversible hydration of carbon dioxide (PubMed:11327835, PubMed:11802772, PubMed:11831900, PubMed:12056894, PubMed:12171926, PubMed:1336460, PubMed:14736236, PubMed:15300855, PubMed:15453828, PubMed:15667203, PubMed:15865431, PubMed:16106378, PubMed:16214338, PubMed:16290146, PubMed:16686544, PubMed:16759856, PubMed:16807956, PubMed:17127057, PubMed:17251017, PubMed:17314045, PubMed:17330962, PubMed:17346964, PubMed:17540563, PubMed:17588751, PubMed:17705204, PubMed:18024029, PubMe
CytoplasmCell membrane
Osteopetrosis, autosomal recessive 3
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB3 is associated with renal tubular acidosis, cerebral calcification (marble brain disease) and in some cases with intellectual disability.
Plays a central role in cell adhesion in hematopoietic cells (PubMed:19234463, PubMed:26359933). Acts by activating the integrin beta-1-3 (ITGB1, ITGB2 and ITGB3) (By similarity). Required for integrin-mediated platelet adhesion and leukocyte adhesion to endothelial cells (PubMed:19234460). Required for activation of integrin beta-2 (ITGB2) in polymorphonuclear granulocytes (PMNs) (By similarity) Isoform 2 may act as a repressor of NF-kappa-B and apoptosis
Cell projection, podosome
Leukocyte adhesion deficiency 3
A disorder characterized by recurrent bacterial infections without pus formation, leukocytosis and major bleeding disorders.
Probable phosphoinositide-binding protein involved in protein sorting and membrane trafficking in endosomes. Plays a role in cilium biogenesis through regulation of the transport and the localization of proteins to the cilium. Required for the localization to the cilium of V-ATPase subunit ATP6V1D and ATP6V0D1, and RAB8A. Involved in osteoclast differentiation and therefore bone resorption
CytoplasmEndosome membraneCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Osteopetrosis, autosomal recessive 8
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB8 is clinically characterized by dense bones with no distinction between outer and inner plates, due to extensive encroachment of cortical bone into the medullary space, increased head circumference, broad open fontanelle, frontal bossing, and hepatosplenomegaly. Osteoclasts number is low and their bone resorptive capacity is impaired.
Slowly voltage-gated channel mediating the exchange of chloride ions against protons (PubMed:18449189, PubMed:21527911). Functions as antiporter and contributes to the acidification of the lysosome lumen and may be involved in maintaining lysosomal pH (PubMed:18449189, PubMed:21527911, PubMed:31155284). The CLC channel family contains both chloride channels and proton-coupled anion transporters that exchange chloride or another anion for protons (By similarity). The presence of conserved gating
Lysosome membrane
Osteopetrosis, autosomal recessive 4
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves.
Cytokine that binds to TNFRSF11B/OPG and to TNFRSF11A/RANK. Osteoclast differentiation and activation factor. Augments the ability of dendritic cells to stimulate naive T-cell proliferation. May be an important regulator of interactions between T-cells and dendritic cells and may play a role in the regulation of the T-cell-dependent immune response. May also play an important role in enhanced bone-resorption in humoral hypercalcemia of malignancy (PubMed:22664871). Induces osteoclastogenesis by
Cell membraneCytoplasmSecreted
Osteopetrosis, autosomal recessive 2
A rare genetic disease characterized by abnormally dense bone, due to defective resorption of immature bone. Osteopetrosis occurs in two forms: a severe autosomal recessive form occurring in utero, infancy, or childhood, and a benign autosomal dominant form occurring in adolescence or adulthood. Recessive osteopetrosis commonly manifests in early infancy with macrocephaly, feeding difficulties, evolving blindness and deafness, bone marrow failure, severe anemia, and hepatosplenomegaly. Deafness and blindness are generally thought to represent effects of pressure on nerves. OPTB2 is characterized by paucity of osteoclasts, suggesting a molecular defect in osteoclast development.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
416 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
17 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Osteopetrose maligna autossômica recessiva
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
Pesquisa e ensaios clínicos
4 ensaios clínicos encontrados.
Publicações mais relevantes
Publicações recentes
Severe hypophosphataemia can be an early sign of osteopetrorickets: a case report.
Infantile autosomal-recessive malignant osteopetrosis.
Familial malignant osteopetrosis in children: a case report.
Autosomal malignant osteopetrosis. From diagnosis to therapy.
Osteoclast morphology in autosomal recessive malignant osteopetrosis due to a TCIRG1 gene mutation.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Osteopetrose maligna autossômica recessiva.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Osteopetrose maligna autossômica recessiva
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Severe hypophosphataemia can be an early sign of osteopetrorickets: a case report.
- Infantile autosomal-recessive malignant osteopetrosis.
- Familial malignant osteopetrosis in children: a case report.
- Autosomal malignant osteopetrosis. From diagnosis to therapy.
- Osteoclast morphology in autosomal recessive malignant osteopetrosis due to a TCIRG1 gene mutation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:667(Orphanet)
- MONDO:0019026(MONDO)
- GARD:15012(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Artigo Wikipedia(Wikipedia)
- Q1755568(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
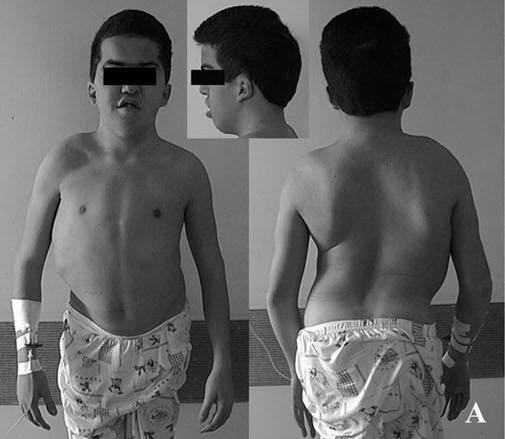
Osteopetrose maligna autossômica recessiva
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA