A Opsismodisplasia é uma doença que afeta os ossos, caracterizada por nanismo (baixa estatura) presente desde o nascimento e características faciais incomuns.
Introdução
O que você precisa saber de cara
A Opsismodisplasia é uma doença que afeta os ossos, caracterizada por nanismo (baixa estatura) presente desde o nascimento e características faciais incomuns.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 27 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 67 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisPhosphatidylinositol (PtdIns) phosphatase that specifically hydrolyzes the 5-phosphate of phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3) to produce PtdIns(3,4)P2, thereby negatively regulating the PI3K (phosphoinositide 3-kinase) pathways (PubMed:16824732). Required for correct mitotic spindle orientation and therefore progression of mitosis (By similarity). Plays a central role in regulation of PI3K-dependent insulin signaling, although the precise molecular mechanisms and signaling
Cytoplasm, cytosolCytoplasm, cytoskeletonMembraneCell projection, filopodiumCell projection, lamellipodiumBasal cell membraneNucleusNucleus speckleCytoplasm, cytoskeleton, spindle pole
Variantes genéticas (ClinVar)
85 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 55 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Opsismodisplasia
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Pamidronate Treatment of a Patient with Opsismodysplasia and a Novel INPPL1 Variant: Efficacy, Mechanism, and Clinical Outcomes.
Opsismodysplasia (OPS) is a rare skeletal dysplasia characterized by delayed bone maturation, distinctive skeletal deformities, and severe growth impairment. Mutations in the INPPL1 gene, particularly homozygous variants, are the primary genetic cause of this condition. While bisphosphonate therapy has shown efficacy in OPS cases with hypophosphatemic rickets, its role in cases without this complication remains unclear. A 2-year-and-2-month-old girl with OPS was treated with intravenous pamidronate (0.5 mg/kg/3 months). Initial evaluations showed severe short stature and low bone mineral density (DEXA SDS: -3.16). After three courses of treatment, the patient achieved independent walking, and her DEXA SDS improved to -2.5 over 1 year. Pamidronate is effective in treating OPS even in the absence of hypophosphatemic rickets, showing potential as a therapeutic option for this rare condition. INPPL1-related opsismodysplasia is characterized by prenatal-onset short stature, short, bowed limbs, characteristic facial features (relative macrocephaly, prominent forehead, midface retrusion, depressed nasal bridge, short nose, anteverted nares, relatively long philtrum), narrow thorax, small hands and feet, delayed epiphyseal mineralization, metaphyseal cupping, and platyspondyly. Complications include increased risk of fractures, cervical spine abnormalities, scoliosis, bone pain, respiratory issues, and delayed gross motor skills. Some individuals have cardiac or kidney manifestations. Prognosis is variable with perinatal demise in some infants. The diagnosis of INPPL1-related opsismodysplasia is established in a proband with characteristic clinical and radiographic features and biallelic pathogenic variants in INPPL1 identified by molecular genetic testing. Targeted therapy: Intravenous bisphosphonate therapy has improved bone mineral density and gross motor function in two individuals with INPPL1-related opsismodysplasia. Supportive care: Cervical spine complications should be managed by specialists familiar with skeletal dysplasias involving the spine including an orthopedist and neurosurgeon; surgical stabilization should be performed to prevent progressive myelopathy; management of scoliosis per orthopedist with surgery when indicated; management of hypophosphatemia, renal phosphate wasting, and bone demineralization per endocrinologist; treatment of respiratory insufficiency per pulmonologist; vaccines to prevent respiratory illnesses; CPAP and surgical management as needed for sleep apnea; feeding therapy with modification of fluid or food texture as needed for swallowing difficulties; aerodigestive evaluation for endoscopy and surgery as needed; management of cardiac manifestations per cardiologist; management of renal manifestations per nephrologist and/or urologist; amplification/hearing device and/or surgery when indicated for hearing impairment; social work and family support. Surveillance: Flexion/extension cervical spine MRI every three months until cervical instability can be excluded in those with instability, risk of cervical cord compression, or limited radiograph interpretation; then MRI every two to three years, preoperatively, and when indicated. Clinical examination for scoliosis every six to 12 months with radiographs when indicated; endocrinology evaluation for hypophosphatemia and renal phosphate wasting every six to 12 months and when indicated; DXA scan when indicated; pulmonary function studies, chest radiographs, swallowing evaluation, and sleep study every six to 12 months and when indicated per pulmonologist; swallowing evaluation as indicated to evaluate for aspiration; developmental assessment to assess gross motor skills annually or as needed; rehabilitation medicine, physical therapy, and occupational therapy consultations when indicated to evaluate function and need for adaptive devices and to support activities of daily living and mobility; clinical cardiac examination with frequency per cardiologist; audiology evaluation annually or as needed; ENT and orthodontic evaluations as needed; assess family and social work needs at each visit. Agents/circumstances to avoid: Individuals with cervical spine instability or who are at risk for cervical spine instability should avoid extreme neck flexion and extension, contact sports, and other at-risk activities. Individuals with bone demineralization should avoid contact sports and other activities associated with an increased risk for fractures. Evaluation of relatives at risk: It is appropriate to clarify the genetic status of apparently asymptomatic at-risk sibs of an affected individual in order to identify as early as possible those who would benefit from prompt initiation of targeted therapy. INPPL1-related opsismodysplasia is inherited in an autosomal recessive manner. If both parents are known to be heterozygous for an INPPL1 pathogenic variant, each sib of an affected individual has at conception a 25% chance of being affected, a 50% chance of being an asymptomatic carrier, and a 25% chance of being unaffected and not a carrier. Once the INPPL1 pathogenic variants have been identified in an affected family member, carrier testing for at-risk relatives and prenatal/preimplantation genetic testing are possible.
Cell expansion for notochord mechanics and endochondral bone lengthening in zebrafish depends on the 5'-inositol phosphatase Inppl1a.
Cell size is a key contributor to tissue morphogenesis. As a notable example, growth plate hypertrophic chondrocytes use cellular biogenesis and disproportionate fluid uptake to expand 10 to 20 times in size to drive lengthening of endochondral bone. Similarly, notochord vacuolated cells expand to one of the largest cell types in the developing embryo to drive axial extension. In zebrafish, the notochord vacuolated cells undergo vacuole fusion to form a single large, fluid-filled vacuole that fills the cytoplasmic space and contributes to vacuolated cell expansion. When this process goes awry, the notochord lacks sufficient hydrostatic pressure to support vertebral bone deposition, resulting in adult spines with misshapen vertebral bones and scoliosis. However, it remains unclear whether endochondral bone and the notochord share common genetic and cellular mechanisms for regulating cell and tissue expansion. Here, we demonstrate that the 5'-inositol phosphatase gene, inppl1a, regulates notochord expansion independent of vacuole fusion, thereby genetically decoupling these processes. We demonstrate that inppl1a-dependent vacuolated cell expansion is essential to establish normal mechanical properties of the notochord and to facilitate the development of a straight spine. Finally, we find that inppl1a is also important for hypertrophic chondrocyte differentiation and endochondral bone lengthening in fish, as has been shown in the human INPPL1-related endochondral bone disorder, opsismodysplasia. Overall, this work reveals a shared mechanism of cell size regulation that influences disparate tissues critical for skeletal development and short-stature disorders.
A Case of Opsismodysplasia with a Novel INPPL1 Variant.
Opsismodysplasia is a rare autosomal recessive genetic skeletal disorder characterized by short stature, short limbs, small hands and feet, delayed bone age, severe platyspondyly, metaphyseal cupping, and facial dysmorphism. Opsismodysplasia is caused by biallelic variants in the INPPL1 gene. Only 38 patients with a confirmed molecular diagnosis have been reported so far. We present a 9-month-old male patient who was referred to our clinic with a suspicion of mucopolysaccharidoses due to facial features and radiographic findings, but urine glycosaminoglycans were within normal ranges. Audiologic and ophthalmologic assessments, transfontanelle ultrasound, and echocardiography were all normal. A renal cortical cyst with a diameter of 33 × 28 mm was detected in abdominal ultrasound. He had dysmorphic findings including relative macrocephaly, midface hypoplasia, depressed nasal bridge, anteverted nostrils, long philtrum, small hands and feet, and brachydactyly. His length was 63 cm (-3.7 SD) and his arm span was 58 cm. Delayed bone age, short metacarpals and phalanges, wide and irregular metaphysis, platyspondyly, anterior beaking of the vertebrae, T12 vertebral hypoplasia, and acetabular dysplasia were noted on X-rays. Exome sequencing revealed a novel homozygous c.147C>G (p.Ser49Arg) variant in INPLL1. Opsismodysplasia is an extremely rare skeletal disorder, and with this case, we further expand the clinical and molecular spectrum of opsismodysplasia.
The Functional Roles of the Src Homology 2 Domain-Containing Inositol 5-Phosphatases SHIP1 and SHIP2 in the Pathogenesis of Human Diseases.
The src homology 2 domain-containing inositol 5-phosphatases SHIP1 and SHIP2 are two proteins involved in intracellular signaling pathways and have been linked to the pathogenesis of several diseases. Both protein paralogs are well known for their involvement in the formation of various kinds of cancer. SHIP1, which is expressed predominantly in hematopoietic cells, has been implicated as a tumor suppressor in leukemogenesis especially in myeloid leukemia, whereas SHIP2, which is expressed ubiquitously, has been implicated as an oncogene in a wider variety of cancer types and is suggested to be involved in the process of metastasis of carcinoma cells. However, there are numerous other diseases, such as inflammatory diseases as well as allergic responses, Alzheimer's disease, and stroke, in which SHIP1 can play a role. Moreover, SHIP2 overexpression was shown to correlate with opsismodysplasia and Alzheimer's disease, as well as metabolic diseases. The SHIP1-inhibitor 3-α-aminocholestane (3AC), and SHIP1-activators, such as AQX-435 and AQX-1125, and SHIP2-inhibitors, such as K161 and AS1949490, have been developed and partly tested in clinical trials, which indicates the importance of the SHIP-paralogs as possible targets in the therapy of those diseases. The aim of this article is to provide an overview of the current knowledge about the involvement of SHIP proteins in the pathogenesis of cancer and other human diseases and to create awareness that SHIP1 and SHIP2 are more than just tumor suppressors and oncogenes.
A conserved regulation of cell expansion underlies notochord mechanics, spine morphogenesis, and endochondral bone lengthening.
Cell size is a key contributor to tissue morphogenesis 1 . As a notable example, growth plate hypertrophic chondrocytes use cellular biogenesis and disproportionate fluid uptake to expand 10-20 times in size to drive lengthening of endochondral bone 2,3 . Similarly, notochordal cells expand to one of the largest cell types in the developing embryo to drive axial extension 4-6 . In zebrafish, the notochord vacuolated cells undergo vacuole fusion to form a single large, fluid-filled vacuole that fills the cytoplasmic space and contributes to vacuolated cell expansion 7 . When this process goes awry, the notochord lacks sufficient hydrostatic pressure to support vertebral bone deposition resulting in adult spines with misshapen vertebral bones and scoliosis 8 . However, it remains unclear whether endochondral bone and the notochord share common genetic and cellular mechanisms for regulating cell and tissue expansion. Here, we demonstrate that the 5'-inositol phosphatase gene, inppl1a , regulates notochord expansion, spine morphogenesis, and endochondral bone lengthening in zebrafish. Furthermore, we show that inppl1a regulates notochord expansion independent of vacuole fusion, thereby genetically decoupling these processes. We demonstrate that inppl1a -dependent notochord expansion is essential to establish normal mechanical properties of the notochord to facilitate the development of a straight spine. Finally, we find that inppl1a is also important for endochondral bone lengthening in fish, as has been shown in the human INPPL1 -related endochondral bone disorder, Opsismodysplasia 9 . Overall, this work reveals a conserved mechanism of cell size regulation that influences disparate tissues critical for skeletal development and short-stature disorders.
Publicações recentes
Pamidronate Treatment of a Patient with Opsismodysplasia and a Novel INPPL1 Variant: Efficacy, Mechanism, and Clinical Outcomes.
INPPL1-Related Opsismodysplasia.
Cell expansion for notochord mechanics and endochondral bone lengthening in zebrafish depends on the 5'-inositol phosphatase Inppl1a.
A Case of Opsismodysplasia with a Novel INPPL1 Variant.
A conserved regulation of cell expansion underlies notochord mechanics, spine morphogenesis, and endochondral bone lengthening.
📚 EuropePMC24 artigos no totalmostrando 13
Pamidronate Treatment of a Patient with Opsismodysplasia and a Novel INPPL1 Variant: Efficacy, Mechanism, and Clinical Outcomes.
Molecular syndromologyCell expansion for notochord mechanics and endochondral bone lengthening in zebrafish depends on the 5'-inositol phosphatase Inppl1a.
Current biology : CBA Case of Opsismodysplasia with a Novel INPPL1 Variant.
Molecular syndromologyA conserved regulation of cell expansion underlies notochord mechanics, spine morphogenesis, and endochondral bone lengthening.
bioRxiv : the preprint server for biologyThe Functional Roles of the Src Homology 2 Domain-Containing Inositol 5-Phosphatases SHIP1 and SHIP2 in the Pathogenesis of Human Diseases.
International journal of molecular sciencesPrenatal-onset INPPL1-related skeletal dysplasia in two unrelated families: Diagnosis and prediction of lethality.
Clinical case reportsPhosphoinositide 5-phosphatases SKIP and SHIP2 in ruffles, the endoplasmic reticulum and the nucleus: An update.
Advances in biological regulationAltered chondrocyte differentiation, matrix mineralization and MEK-Erk1/2 signaling in an INPPL1 catalytic knock-out mouse model of opsismodysplasia.
Advances in biological regulationFibroblasts derived from patients with opsismodysplasia display SHIP2-specific cell migration and adhesion defects.
Human mutationSHIP2: Structure, Function and Inhibition.
Chembiochem : a European journal of chemical biologyINPPL1 gene mutations in opsismodysplasia.
Journal of human geneticsNovel compound heterozygous mutations in inositol polyphosphate phosphatase-like 1 in a family with severe opsismodysplasia.
Clinical dysmorphologyOpsismodysplasia: Phosphate Wasting Osteodystrophy Responds to Bisphosphonate Therapy.
Frontiers in pediatricsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Opsismodisplasia.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Opsismodisplasia
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Pamidronate Treatment of a Patient with Opsismodysplasia and a Novel INPPL1 Variant: Efficacy, Mechanism, and Clinical Outcomes.
- Cell expansion for notochord mechanics and endochondral bone lengthening in zebrafish depends on the 5'-inositol phosphatase Inppl1a.
- A Case of Opsismodysplasia with a Novel INPPL1 Variant.
- The Functional Roles of the Src Homology 2 Domain-Containing Inositol 5-Phosphatases SHIP1 and SHIP2 in the Pathogenesis of Human Diseases.
- A conserved regulation of cell expansion underlies notochord mechanics, spine morphogenesis, and endochondral bone lengthening.
- INPPL1-Related Opsismodysplasia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:2746(Orphanet)
- OMIM OMIM:258480(OMIM)
- MONDO:0009785(MONDO)
- GARD:4098(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q7098730(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
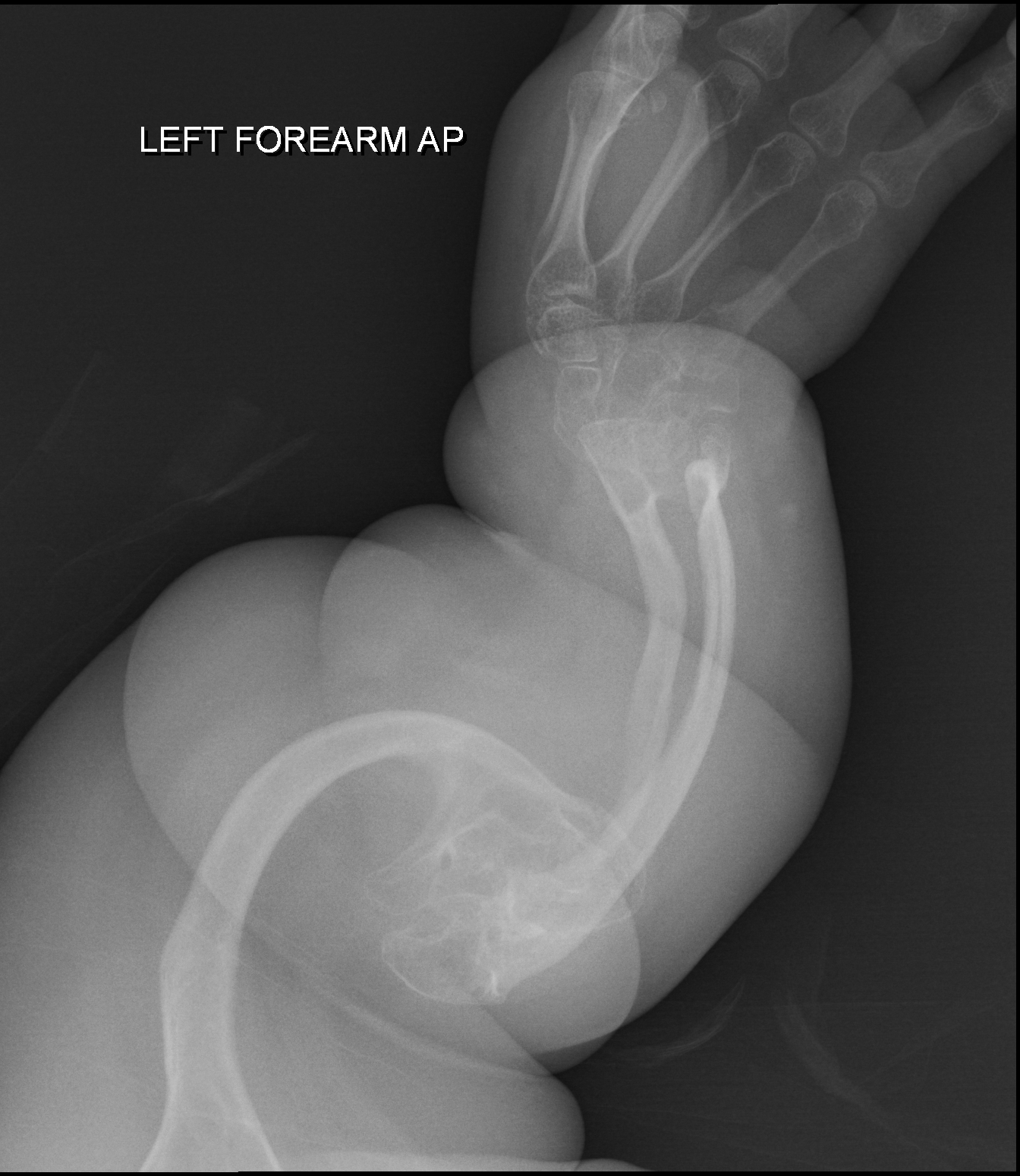
Opsismodisplasia
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata