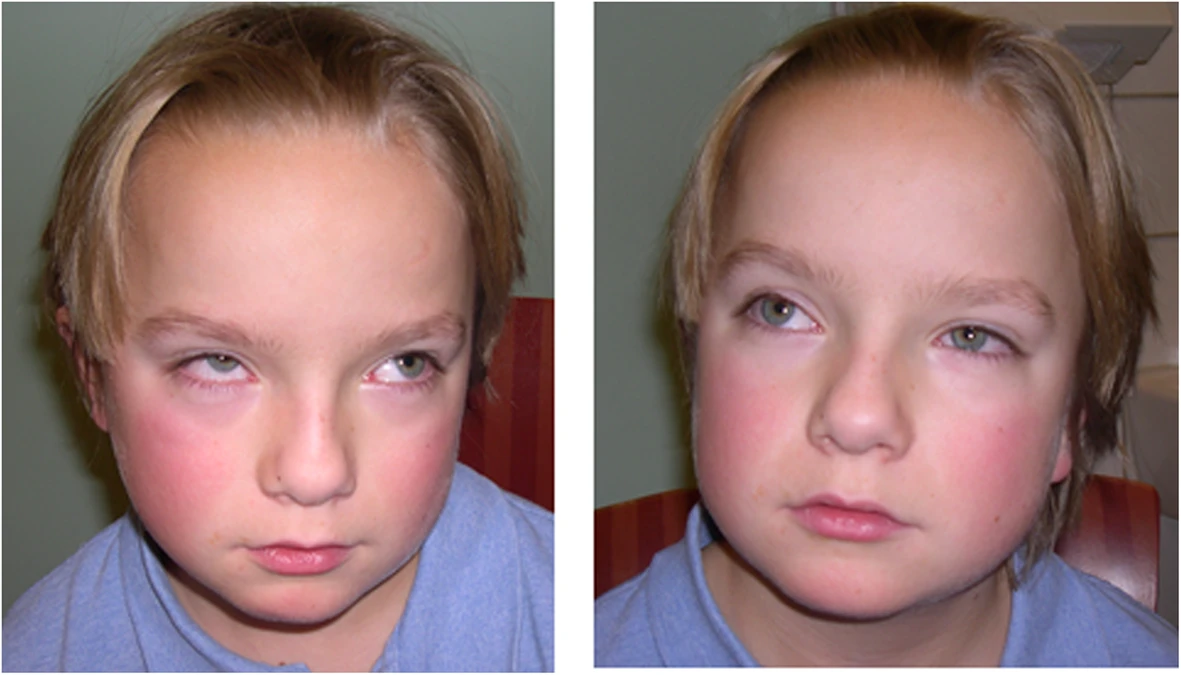
A síndrome de Joubert (JS) e distúrbios relacionados (JSRD) são um grupo de síndromes de atraso no desenvolvimento / anomalias congênitas múltiplas em que a característica obrigatória é o "sinal do dente molar" (MTS), uma malformação complexa do mesencéfalo-rombencéfalo reconhecível na imagem cerebral. O MTS é caracterizado por hipodisplasia do verme cerebelar, espessamento e desorientação dos pedúnculos cerebelares superiores e fossa interpeduncular anormalmente profunda.
Introdução
O que você precisa saber de cara
A síndrome de Joubert (JS) e distúrbios relacionados (JSRD) são um grupo de síndromes de atraso no desenvolvimento / anomalias congênitas múltiplas em que a característica obrigatória é o "sinal do dente molar" (MTS), uma malformação complexa do mesencéfalo-rombencéfalo reconhecível na imagem cerebral. O MTS é caracterizado por hipodisplasia do verme cerebelar, espessamento e desorientação dos pedúnculos cerebelares superiores e fossa interpeduncular anormalmente profunda.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 83 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 256 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
16 genes identificados com associação a esta condição. Padrão de herança: Autosomal recessive, X-linked recessive.
Required during ciliogenesis for tubulin glutamylation in cilium. Probably acts by participating in the transport of TTLL6, a tubulin polyglutamylase, between the basal body and the cilium
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 15
An autosomal recessive disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis and polydactyly.
Plays a role in the microtubule-dependent coupling of the nucleus and the centrosome. Involved in the processes that regulate centrosome-mediated interkinetic nuclear migration (INM) of neural progenitors and for proper positioning of neurons during brain development. Also implicated in the migration and selfrenewal of neural progenitors. Required for centriole duplication and maturation during mitosis and subsequent ciliogenesis (By similarity). Required for the recruitment of CEP295 to the pro
Cytoplasm, cytoskeleton, microtubule organizing center, centrosome
Short-rib thoracic dysplasia 13 with or without polydactyly
A form of short-rib thoracic dysplasia, a group of autosomal recessive ciliopathies that are characterized by a constricted thoracic cage, short ribs, shortened tubular bones, and a 'trident' appearance of the acetabular roof. Polydactyly is variably present. Non-skeletal involvement can include cleft lip/palate as well as anomalies of major organs such as the brain, eye, heart, kidneys, liver, pancreas, intestines, and genitalia. Some forms of the disease are lethal in the neonatal period due to respiratory insufficiency secondary to a severely restricted thoracic cage, whereas others are compatible with life. Disease spectrum encompasses Ellis-van Creveld syndrome, asphyxiating thoracic dystrophy (Jeune syndrome), Mainzer-Saldino syndrome, and short rib-polydactyly syndrome.
Phosphatidylinositol (PtdIns) phosphatase that specifically hydrolyzes the 5-phosphate of phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3), phosphatidylinositol 4,5-bisphosphate (PtdIns(4,5)P2) and phosphatidylinositol 3,5-bisphosphate (PtdIns(3,5)P2) (By similarity) (PubMed:10764818). Specific for lipid substrates, inactive towards water soluble inositol phosphates (PubMed:10764818). Plays an essential role in the primary cilium by controlling ciliary growth and phosphoinositide 3-kin
Cytoplasm, cytoskeleton, cilium axonemeGolgi apparatus, Golgi stack membraneCell membraneCell projection, ruffleCytoplasmNucleus
Joubert syndrome 1
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Transcription factor that can both act as an activator or a repressor depending on the context. Plays a central role in BMP signaling and olfactory neurogenesis. Associates with SMADs in response to BMP2 leading to activate transcription of BMP target genes. Acts as a transcriptional repressor via its interaction with EBF1, a transcription factor involved in terminal olfactory receptor neurons differentiation; this interaction preventing EBF1 to bind DNA and activate olfactory-specific genes. In
Nucleus
Nephronophthisis 14
An autosomal recessive disorder manifesting as infantile-onset kidney disease, cerebellar vermis hypoplasia, and situs inversus. Nephronophthisis is a progressive tubulo-interstitial kidney disorder histologically characterized by modifications of the tubules with thickening of the basement membrane, interstitial fibrosis and, in the advanced stages, medullary cysts.
Essential for primary ciliogenesis and embryonic development, facilitating the activation of Hedgehog (Hh) signaling pathway. Disrupts the interaction of GLI2 and GLI3 with the negative regulator SUFU. Inhibiting SUFU's interaction with GLI2 promotes the entry of GLI2 into the nucleus, allowing it to activate Hh target gene expression. Disrupting SUFU's interaction with GLI3 prevents its conversion into the repressor form, leading to increased nuclear GLI3 and enhanced Hh signaling. Required for
MembraneCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 2
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for ciliogenesis and sonic hedgehog/SHH signaling (By similarity)
CytoplasmCytoplasm, cytoskeleton, cilium basal body
Meckel syndrome 6
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Required for ciliary structure and function. Part of the tectonic-like complex which is required for tissue-specific ciliogenesis and may regulate ciliary membrane composition (By similarity). Involved in centrosome migration to the apical cell surface during early ciliogenesis. Involved in the regulation of cilia length and appropriate number through the control of centrosome duplication. Is a key regulator of stereociliary bundle orientation (By similarity). Required for epithelial cell branch
Cell membraneEndoplasmic reticulum membraneCell projection, ciliumCytoplasm, cytoskeleton, cilium basal body
Negatively regulates signaling through the G-protein coupled thromboxane A2 receptor (TBXA2R) (PubMed:19464661). May be involved in mechanisms like programmed cell death, craniofacial development, patterning of the limbs, and formation of the left-right axis (By similarity). Involved in the organization of apical junctions; the function is proposed to implicate a NPHP1-4-8 module. Does not seem to be strictly required for ciliogenesis (PubMed:19464661). Involved in establishment of planar cell p
CytoplasmCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, cilium axonemeCytoplasm, cytoskeleton, microtubule organizing center, centrosomeCell junction, tight junction
Required for ciliogenesis and sonic hedgehog/SHH signaling. Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1. May play a role in early ciliogenesis in the disappearance of centriolar satellites that preceeds ciliary vesicle formation (PubMed:24421332). Involved in regulation of cell intracellular organization. Involved in regulation of cell polarity (By similarity). Required for asymmetrical localization of CEP
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomePhotoreceptor inner segmentCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasm, cytoskeleton, cilium basal body
Joubert syndrome 23
A mild form of Joubert syndrome, a disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Involved in vesicle trafficking and required for ciliogenesis, formation of primary non-motile cilium, and recruitment of RAB8A to the basal body of primary cilium. Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Involved in neuronal differentiation. As a positive modulator of classical Wnt signaling, may play a crucial role in cili
Cytoplasm, cytoskeleton, cilium basal bodyCell junction, adherens junctionCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriole
Joubert syndrome 3
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. Joubert syndrome type 3 shows minimal extra central nervous system involvement and appears not to be associated with renal dysfunction.
Required for ciliogenesis
Vacuole membraneCell projection, cilium
Joubert syndrome 16
An autosomal recessive disorder characterized by oculomotor apraxia, variable coloboma, and rare kidney involvement. Neuroradiologically, it is characterized by an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and polydactyly.
Involved in early and late steps in cilia formation. Its association with CCP110 is required for inhibition of primary cilia formation by CCP110 (PubMed:18694559). May play a role in early ciliogenesis in the disappearance of centriolar satellites and in the transition of primary ciliar vesicles (PCVs) to capped ciliary vesicles (CCVs). Required for the centrosomal recruitment of RAB8A and for the targeting of centriole satellite proteins to centrosomes such as of PCM1 (PubMed:24421332). Require
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centriolar satelliteNucleusCell projection, ciliumCytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome, centrioleCytoplasmic vesicle
Joubert syndrome 5
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease. Joubert syndrome type 5 shares the neurologic and neuroradiologic features of Joubert syndrome together with severe retinal dystrophy and/or progressive renal failure characterized by nephronophthisis.
Component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Involved in centrosome migration to the apical cell surface during early ciliogenesis. Required for ciliary structure and function, including a role in regulating length and appropriate number through modulating centrosome duplication. Required for cell branching morphology
Cytoplasm, cytoskeleton, cilium basal bodyCytoplasm, cytoskeleton, microtubule organizing center, centrosome
Meckel syndrome 1
A disorder characterized by a combination of renal cysts and variably associated features including developmental anomalies of the central nervous system (typically encephalocele), hepatic ductal dysplasia and cysts, and polydactyly.
Transmembrane component of the tectonic-like complex, a complex localized at the transition zone of primary cilia and acting as a barrier that prevents diffusion of transmembrane proteins between the cilia and plasma membranes. Required for ciliogenesis and sonic hedgehog/SHH signaling (By similarity)
Cell projection, cilium membrane
Joubert syndrome 20
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy and renal disease.
Component of the transition zone in primary cilia. Required for ciliogenesis
MembraneCell projection, cilium
Joubert syndrome 14
An autosomal recessive disorder characterized by severe intellectual disability, hypotonia, breathing abnormalities in infancy, and dysmorphic facial features. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include renal disease, abnormal eye movements, and postaxial polydactyly.
May play a role in cell-cycle-dependent microtubule organization
Cytoplasm, cytoskeleton, microtubule organizing center, centrosomeCytoplasm, cytoskeleton, spindleCytoplasm, cytoskeleton, spindle poleCell projection, cilium
Joubert syndrome 21
A disorder presenting with cerebellar ataxia, oculomotor apraxia, hypotonia, neonatal breathing abnormalities and psychomotor delay. Neuroradiologically, it is characterized by cerebellar vermian hypoplasia/aplasia, thickened and reoriented superior cerebellar peduncles, and an abnormally large interpeduncular fossa, giving the appearance of a molar tooth on transaxial slices (molar tooth sign). Additional variable features include retinal dystrophy, renal disease, liver fibrosis, and polydactyly.
Variantes genéticas (ClinVar)
405 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 243 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
12 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome Joubert e doenças relacionadas
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
Challenges in postoperative management of a patient with primary ciliary dyskinesia and Joubert syndrome and related disorders with congenital heart disease.
We report the case of a male infant born at 37 weeks and 4 days of gestation with a birth weight of 3396 g. He presented with hypotonia, congenital heart disease, heterotaxy and polydactyly. Based on physical examination and brain MRI, which revealed the characteristic molar tooth sign, he was diagnosed with Joubert syndrome and related disorders (JSRD). He experienced repeated episodes of atelectasis, making extubation challenging. Electron microscopy revealed ciliary structural abnormalities, leading to the diagnosis of primary ciliary dyskinesia as well as JSRD. Despite multiple medical interventions and two surgical procedures for heart failure, he died at 10 months old due to progressing chronic heart failure and pulmonary infections.This report presents the course leading to the diagnosis of ciliary dysfunction-associated multiple malformations, based on the diverse symptoms. Furthermore, we discuss the potential risk of postoperative complications associated with ciliary dysfunction.
Persistent left superior vena cava discovered during central line insertion in a patient with Joubert syndrome: a case report.
Persistent left superior vena cava is the most common thoracic venous anomaly. It is usually asymptomatic and discovered incidentally during diagnostic or therapeutic interventions. It can complicate central line insertion, causing arrythmias and thromboembolic complications. Joubert syndrome and related disorders are a group of rare congenital disorders associated with developmental abnormalities and multiple organ system involvement including renal, hepatic, and neurological manifestations. We report a case of a 12-year-old Kuwaiti Arab male child with Joubert syndrome, who was diagnosed with renal and hepatic failure and required central line insertion to initiate total parenteral nutrition. The central line was inserted through the left internal jugular vein because the patient had a dialysis catheter inserted in the right subclavian vein. After the line insertion, a chest X-ray revealed that the catheter appeared to descend directly into the left mediastinum rather than crossing to the right mediastinum through the innominate vein. Blood gas sample and subsequent echocardiography confirmed the presence of the catheter in a persistent left superior vena cava. The catheter was kept in place and used without complication for total parenteral nutrition and resuscitation for 18 days. Although persistent left superior vena cava is a rare anomaly, knowledge of its presence is important for the clinician in order to avoid complications associated with utilizing it for venous access, and to avoid unnecessary replacement for an uncomplicated catheter inserted in it. This is especially important for patients who would be expected to require invasive interventions, such as patients with Joubert syndrome.
Nonequivalence of Zfp423 premature termination codons in mice.
Genetic variants that introduce a premature termination codon (PTC) are often assumed equivalent and functionally null. Exceptions depend on the specific architectures of the affected mRNA and protein. Here we address phenotypic differences among early truncating variants of mouse Zfp423, whose phenotypes resemble Joubert Syndrome and Related Disorders. We replicate quantitative differences previously seen between presumptive null PTC variants based on their position in the coding sequence. We show with reciprocal congenic strains that large phenotype differences between two PTC variants with the same predicted stop and reinitiation codons are due to the specific allele rather than different strain backgrounds, with no evidence for induced exon skipping. Differences in RNA structure, however, could influence translation rate across the affected exon. Using a reporter assay, we find differences in translational reinitiation between 2 deletion variants that correlate with predicted RNA structure rather than distance from the canonical initiation codon. These results confirm and extend earlier evidence for differences among Zfp423 PTC variants, identify parameters for translational reinitiation after an early termination codon, and reinforce caution in the null interpretation of early PTC variants.
Nonequivalence of Zfp423 premature termination codons in mice.
Genetic variants that introduce a premature termination codon (PTC) are often assumed equivalent and functionally null. Exceptions depend on the specific architectures of the affected mRNA and protein. Here we address phenotypic differences among early truncating variants of mouse Zfp423, whose phenotypes resemble Joubert Syndrome and Related Disorders (JSRD). We replicate quantitative differences previously seen between presumptive null PTC variants based on their position in the coding sequence. We show with reciprocal congenic strains that large phenotype differences between two PTC variants with the same predicted stop and reinitiation codons is due to the specific allele rather than different strain backgrounds, with no evidence for induced exon skipping. Differences in RNA structure, however, could influence translation rate across the affected exon. Using a reporter assay, we find differences in translational reinitiation between two deletion variants that corelate with predicted RNA structure rather than distance from the canonical initiation codon. These results confirm and extend earlier evidence for differences among Zfp423 PTC variants, identify parameters for translational reinitiation after an early termination codon, and reinforce caution in the null interpretation of early PTC variants.
Neuroimaging Characteristics as Diagnostic Tools in Joubert Syndrome and Related Disorders: A Case Report and Literature Review.
Joubert syndrome and related disorders (JSRD) present diagnostic challenges due to their varied clinical features. Neuroimaging, particularly MRI and CT, is critical for identifying the distinctive "molar tooth sign" and other neuroanatomical abnormalities. This case report and literature review emphasize the role of neuroimaging in diagnosing JSRD. Our search targeted pediatric cases with terms like "Joubert anomaly" and "diagnostic imaging." Key findings include cerebellar vermal agenesis, ataxia, developmental delay, and oculomotor apraxia. Cognitive impairment ranges widely, complicating assessment. CT scans reveal dysplastic or absent cerebellar vermis, while MRI shows the characteristic "molar tooth" sign and additional abnormalities such as malformed cerebellar peduncles and enlarged posterior fossa. Accurate diagnosis of JSRD depends on correlating clinical symptoms with specific radiological findings. A multidisciplinary approach is vital for managing this complex disorder.
Publicações recentes
Clinical, genetic and bioinformatic analysis of Saudi families with Joubert syndrome and related disorders.
Case Report: A case of Joubert syndrome in twin pregnancy: MRI manifestations and literature review.
Challenges in postoperative management of a patient with primary ciliary dyskinesia and Joubert syndrome and related disorders with congenital heart disease.
Persistent left superior vena cava discovered during central line insertion in a patient with Joubert syndrome: a case report.
📚 EuropePMC32 artigos no totalmostrando 30
Challenges in postoperative management of a patient with primary ciliary dyskinesia and Joubert syndrome and related disorders with congenital heart disease.
BMJ case reportsPersistent left superior vena cava discovered during central line insertion in a patient with Joubert syndrome: a case report.
Journal of medical case reportsNonequivalence of Zfp423 premature termination codons in mice.
GeneticsNeuroimaging Characteristics as Diagnostic Tools in Joubert Syndrome and Related Disorders: A Case Report and Literature Review.
CureusPontine tegmental cap dysplasia: the role of diffusion tensor imaging.
BMJ case reportsRehabilitation Approach for Children With Joubert Syndrome and Related Disorders.
CureusSuspected Autosomal Recessive Polycystic Kidney Disease but Cerebellar Vermis Hypoplasia, Oligophrenia Ataxia, Coloboma, and Hepatic Fibrosis (COACH) Syndrome in Retrospect, A Delayed Diagnosis Aided by Genotyping and Reverse Phenotyping: A Case Report and A Review of the Literature.
NephronExpression patterns of ciliopathy genes ARL3 and CEP120 reveal roles in multisystem development.
BMC developmental biologyRE: Clinical and Molecular Diagnosis of Joubert Syndrome and Related Disorders.
Pediatric neurologyClinical and Molecular Diagnosis of Joubert Syndrome and Related Disorders.
Pediatric neurologyHealthcare recommendations for Joubert syndrome.
American journal of medical genetics. Part AEarly and Severe Polycystic Kidney Disease and Related Ciliopathies: An Emerging Field of Interest.
NephronFourth ventricle index: sonographic marker for severe fetal vermian dysgenesis/agenesis.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyPrenatal diagnosis of Joubert syndrome: A case report and literature review.
MedicineA Homozygous Missense Variant in INPP5E Associated with Joubert Syndrome and Related Disorders.
Molecular syndromologyDecaying molar tooth sign in Joubert syndrome and related disorders is correlated to a displacement of the corticospinal tract.
NeuroradiologyRecent advances in the molecular diagnosis of polycystic kidney disease.
Expert review of molecular diagnosticsFunctional validation of novel MKS3/TMEM67 mutations in COACH syndrome.
Scientific reportsIn Vitro Modeling Using Ciliopathy-Patient-Derived Cells Reveals Distinct Cilia Dysfunctions Caused by CEP290 Mutations.
Cell reportsHassab's operation for Joubert syndrome with congenital hepatic fibrosis: A case report.
International journal of surgery case reportsWhole-exome sequencing and digital PCR identified a novel compound heterozygous mutation in the NPHP1 gene in a case of Joubert syndrome and related disorders.
BMC medical geneticsMutations in KIAA0753 cause Joubert syndrome associated with growth hormone deficiency.
Human geneticsRenal cystic disease and associated ciliopathies.
Current opinion in obstetrics & gynecologyMolecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center.
Genetics in medicine : official journal of the American College of Medical GeneticsEnlarged intracranial translucency and molar tooth sign in the first trimester as features of Joubert syndrome and related disorders.
Ultrasound in obstetrics & gynecology : the official journal of the International Society of Ultrasound in Obstetrics and GynecologyAN UNUSUAL PRESENTATION OF JOUBERT SYNDROME AND RELATED DISORDERS IN A NEWBORN: PANHYPOPITUITARISM.
Genetic counseling (Geneva, Switzerland)The diagnostic utility of exome sequencing in Joubert syndrome and related disorders.
Journal of human geneticsJoubert syndrome and related disorders: a rare cause of intrahepatic portal hypertension in childhood.
European review for medical and pharmacological sciencesSyndromic ciliopathies: From single gene to multi gene analysis by SNP arrays and next generation sequencing.
Molecular and cellular probesUncrossed epileptic seizures in Joubert syndrome.
BMJ case reportsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome Joubert e doenças relacionadas.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome Joubert e doenças relacionadas
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Challenges in postoperative management of a patient with primary ciliary dyskinesia and Joubert syndrome and related disorders with congenital heart disease.
- Persistent left superior vena cava discovered during central line insertion in a patient with Joubert syndrome: a case report.
- Nonequivalence of Zfp423 premature termination codons in mice.
- Nonequivalence of Zfp423 premature termination codons in mice.
- Neuroimaging Characteristics as Diagnostic Tools in Joubert Syndrome and Related Disorders: A Case Report and Literature Review.
- Clinical, genetic and bioinformatic analysis of Saudi families with Joubert syndrome and related disorders.
- Case Report: A case of Joubert syndrome in twin pregnancy: MRI manifestations and literature review.
- Joubert Syndrome.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:140874(Orphanet)
- MONDO:0015369(MONDO)
- GARD:19931(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55345933(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Síndrome Joubert e doenças relacionadas
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata