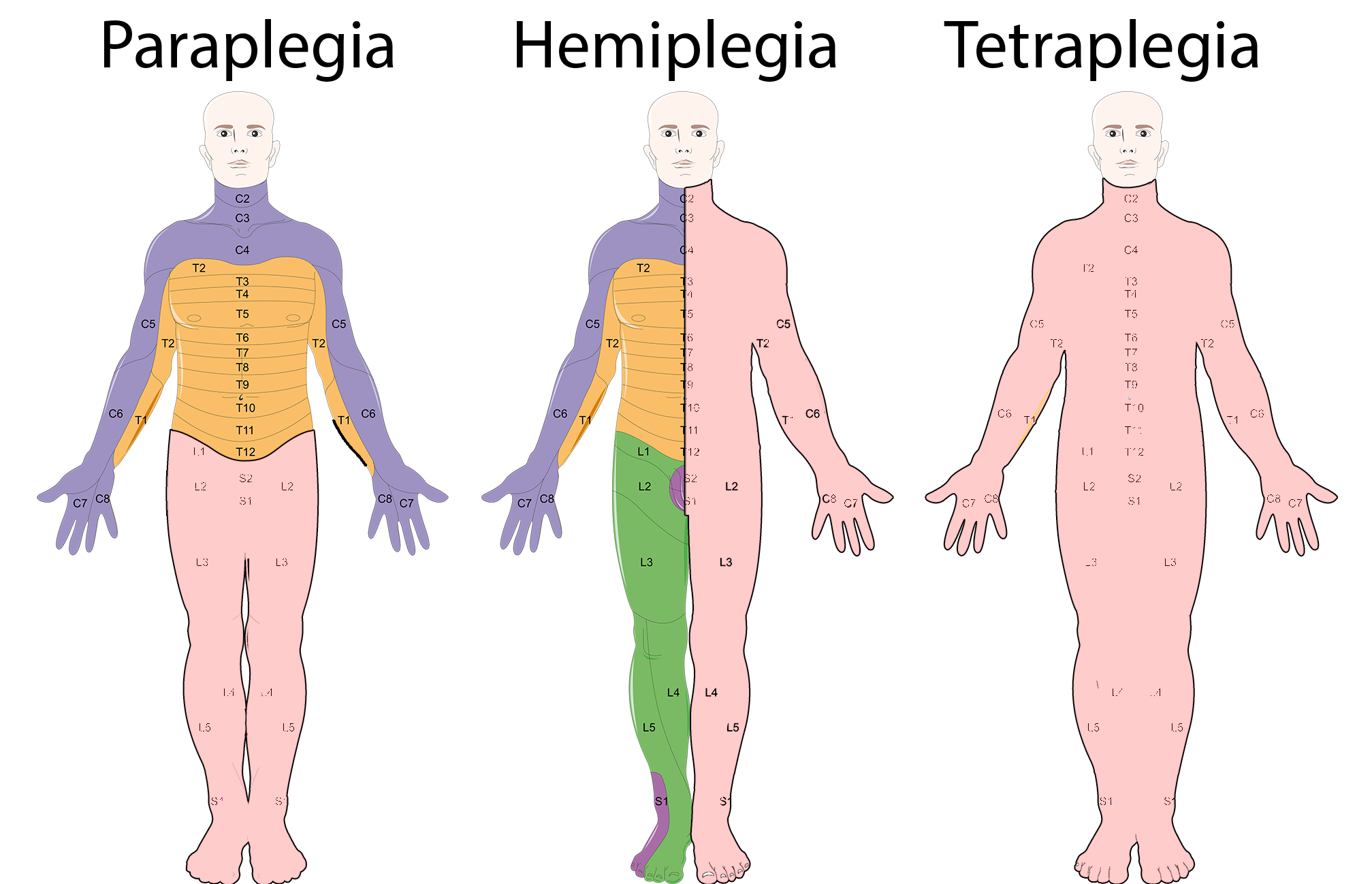
A paraplegia espástica ligada ao X, tipo 34, é uma forma pura de paraplegia espástica hereditária, caracterizada pelo início no final da infância ou no início da vida adulta, com rigidez e fraqueza nas pernas que progridem lentamente, andar rígido, reflexos muito ativos e exagerados nas pernas, e contrações rítmicas e involuntárias no tornozelo (clônus). Dor nas pernas e diminuição da sensação de vibração nas pernas também são relatadas em alguns pacientes mais velhos.
Introdução
O que você precisa saber de cara
A paraplegia espástica ligada ao X, tipo 34, é uma forma pura de paraplegia espástica hereditária, caracterizada pelo início no final da infância ou no início da vida adulta, com rigidez e fraqueza nas pernas que progridem lentamente, andar rígido, reflexos muito ativos e exagerados nas pernas, e contrações rítmicas e involuntárias no tornozelo (clônus). Dor nas pernas e diminuição da sensação de vibração nas pernas também são relatadas em alguns pacientes mais velhos.
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Partes do corpo afetadas
+ 10 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 15 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Paraplegia espástica ligada ao X tipo 34
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
L1 Syndrome Prenatal Diagnosis Supplemented by Functional Analysis of One L1CAM Gene Missense Variant.
L1 syndrome, a complex X-linked neurological disorder, is caused by mutations in the L1 cell adhesion molecule (L1CAM) gene. L1CAM molecule is a member of immunoglobulin (Ig) superfamily of neural cell adhesion molecules (CAMs), which plays a pivotal role in the developing nervous system. In this study, a L1CAM gene exonic missense variant (c.1108G > A, p.G370R) was identified in two induced fetuses (abnormal fetuses), who presented corpus callosum agenesis accompanied with hydrocephalus. Clinical data, published literature, online database, and bioinformatic analysis suggest that the single-nucleotide variant of L1CAM gene is a likely pathogenic mutation. In vitro assays were performed to evaluate the effects of this variant. Based on NSC-34/COS-7 cells transfected with wild-type (L1-WT) and mutated (L1-G370R) plasmids, the L1CAM gene exonic missense variant (c.1108G > A, p.G370R) reduced cell surface expression, induced partial endoplasmic reticulum retention, affected posttranslational modification, and reduced protein's homophilic adhesive ability, but did not induce endoplasmic reticulum stress, which might probably associate with L1 syndrome. Finally, 35 isolated fetuses were screened for L1CAM gene variants by Sanger sequencing. These cases all prenatally suspected of corpus callosum agenesis accompanied with hydrocephalus, which may relate to L1 syndrome. Consequently, one L1CAM gene single missense variant (c.550C > T, p.R184W) was detected in one fetus. Our results provided evidence that the L1CAM gene missense variant (c.1108G > A, p.G370R) may relate to L1 syndrome. The findings of this study suggest a potential possibility of L1CAM gene screening for prenatal diagnoses for fetuses presented corpus callosum agenesis accompanied with hydrocephalus.
Genetic mimics of cerebral palsy.
The term "cerebral palsy mimic" is used to describe a number of neurogenetic disorders that may present with motor symptoms in early childhood, resulting in a misdiagnosis of cerebral palsy. Cerebral palsy describes a heterogeneous group of neurodevelopmental disorders characterized by onset in infancy or early childhood of motor symptoms (including hypotonia, spasticity, dystonia, and chorea), often accompanied by developmental delay. The primary etiology of a cerebral palsy syndrome should always be identified if possible. This is particularly important in the case of genetic or metabolic disorders that have specific disease-modifying treatment. In this article, we discuss clinical features that should alert the clinician to the possibility of a cerebral palsy mimic, provide a practical framework for selecting and interpreting neuroimaging, biochemical, and genetic investigations, and highlight selected conditions that may present with predominant spasticity, dystonia/chorea, and ataxia. Making a precise diagnosis of a genetic disorder has important implications for treatment, and for advising the family regarding prognosis and genetic counseling. © 2019 International Parkinson and Movement Disorder Society.
Publicações recentes
Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia.
Locus and allelic heterogeneity in five families with hereditary spastic paraplegia.
PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus-Merzbacher disease patients.
Pelizaeus-Merzbacher disease and spastic paraplegia type 2: two faces of myelin loss from mutations in the same gene.
Hereditary spastic paraplegia: genetic heterogeneity and genotype-phenotype correlation.
📚 EuropePMC11 artigos no totalmostrando 2
L1 Syndrome Prenatal Diagnosis Supplemented by Functional Analysis of One L1CAM Gene Missense Variant.
Reproductive sciences (Thousand Oaks, Calif.)Genetic mimics of cerebral palsy.
Movement disorders : official journal of the Movement Disorder SocietyAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Paraplegia espástica ligada ao X tipo 34.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Paraplegia espástica ligada ao X tipo 34
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- L1 Syndrome Prenatal Diagnosis Supplemented by Functional Analysis of One L1CAM Gene Missense Variant.
- Genetic mimics of cerebral palsy.Movement disorders : official journal of the Movement Disorder Society· 2019· PMID 30913345mais citado
- Loss-of-function variants in the CAPN1 activator CD99L2 cause X-linked spastic ataxia.
- Locus and allelic heterogeneity in five families with hereditary spastic paraplegia.
- PLP1 gene duplication causes overexpression and alteration of the PLP/DM20 splicing balance in fibroblasts from Pelizaeus-Merzbacher disease patients.
- Pelizaeus-Merzbacher disease and spastic paraplegia type 2: two faces of myelin loss from mutations in the same gene.
- Hereditary spastic paraplegia: genetic heterogeneity and genotype-phenotype correlation.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:171607(Orphanet)
- OMIM OMIM:300750(OMIM)
- MONDO:0010418(MONDO)
- GARD:17063(GARD (NIH))
- Busca completa no PubMed(PubMed)
- Q32142896(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Paraplegia espástica ligada ao X tipo 34
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Indexação biomédica
- fonte: MeSH (NLM)
- Dado público estruturado
- fonte: Wikidata