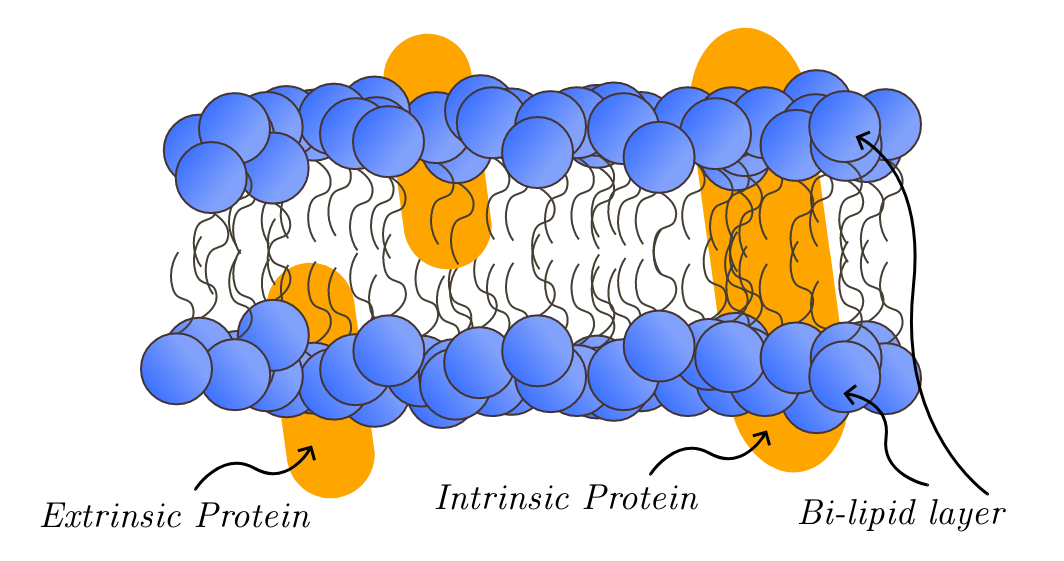
O complexo oligomérico de Golgi conservado, também conhecido como complexo COG, é uma proteína de membrana extrínseca encontrada no aparelho de Golgi em eucariotos. Este complexo é conservado evolutivamente e possui subunidades homólogas encontradas em todas as espécies de eucariotos. O complexo COG foi descoberto pela primeira vez em 1981 em células de ovário de hamster chinês. Essas células de ovário apresentavam mutações nos receptores de lipoproteína de baixa densidade (LDL), o que afetava a função das enzimas de glicosilação do Golgi. No entanto, o complexo COG como um todo não foi totalmente compreendido até 2004, quando uma deficiência em uma das subunidades do COG foi associada a distúrbios congênitos da glicosilação. Essas descobertas levaram à percepção de que o complexo COG desempenha um papel importante na glicosilação e na classificação de proteínas.
Introdução
O que você precisa saber de cara
Visão geral
O Defeito no complexo de Golgi oligomérico conservado é uma doença genética rara que afeta o funcionamento de uma estrutura celular chamada complexo de Golgi. Esse complexo é responsável por modificar, empacotar e enviar proteínas para os lugares certos dentro da célula. Quando há um defeito nesse processo, diversos órgãos e sistemas do corpo podem ser afetados, resultando em uma ampla variedade de sintomas. A condição é causada por alterações (mutações) em genes que produzem as subunidades do complexo de Golgi oligomérico conservado (COG).[1][3]
Sinais e sintomas
Os sinais e sintomas do Defeito no complexo de Golgi oligomérico conservado podem variar muito de pessoa para pessoa, mas frequentemente incluem problemas neurológicos, dificuldades alimentares, infecções recorrentes e alterações na face e nos ossos. Entre os fenótipos (características observáveis) descritos na literatura estão: microcefalia secundária (cabeça pequena que se desenvolve após o nascimento), deficiência intelectual grave, atrofia do músculo esquelético, atrofia cortical temporal, agenesia do vermis cerebelar (parte do cerebelo não se forma), sequência de Pierre-Robin (que pode incluir queixo pequeno, língua para trás e fenda no palato), microtia (orelha pequena ou malformada), filtro labial profundo (sulco entre o nariz e o lábio superior mais marcado), borda do vermelhão espessa (lábios grossos), formato facial anormal, teto acetabular plano e fossa acetabular rasa (alterações no quadril), desvio ulnar do dedo (dedos tortos para o lado do cotovelo), hipertensão arterial pulmonar (pressão alta na artéria do pulmão), dilatação do septo atrial e defeito do septo atrial (problemas no coração), sepse neonatal (infecção generalizada no recém-nascido), infecções respiratórias e gastrointestinais recorrentes, diarreia intermitente, dificuldades alimentares, anormalidade da cascata de coagulação (problemas de coagulação do sangue), atividade reduzida da proteína S (um anticoagulante natural), hipoplasia do esmalte (esmalte dos dentes fino ou ausente) e pele de laranja (aspecto da pele com pequenas depressões).[1][3]
Causas genéticas
A doença é causada por mutações em genes que codificam as subunidades do complexo de Golgi oligomérico conservado (COG). Os genes já identificados como associados a essa condição são: COG1, COG2, COG4, COG5, COG6, COG7 e COG8. Cada um desses genes fornece instruções para a produção de uma peça (subunidade) do complexo COG. Mutações em qualquer um desses genes podem prejudicar a montagem ou a função do complexo, levando aos sintomas da doença. A herança (como a condição é passada de pais para filhos) não está especificada nas fontes disponíveis.[1][4]
Diagnóstico
O diagnóstico do Defeito no complexo de Golgi oligomérico conservado é baseado na avaliação clínica dos sinais e sintomas e confirmado por meio de testes genéticos. Existem 336 testes genéticos disponíveis para essa condição, e 311 variantes (alterações no DNA) já foram registradas no ClinVar, um banco de dados público que reúne informações sobre a relação entre variantes genéticas e doenças. O diagnóstico genético é essencial para diferenciar essa condição de outras doenças raras com sintomas semelhantes.[1][4]
Tratamento e manejo
Atualmente, não há um tratamento específico ou medicamento aprovado para o Defeito no complexo de Golgi oligomérico conservado. O manejo da doença é focado no alívio dos sintomas e no suporte às necessidades individuais de cada paciente. Isso pode incluir acompanhamento com equipe multidisciplinar (neurologista, geneticista, fisioterapeuta, fonoaudiólogo, nutricionista, entre outros), intervenções para dificuldades alimentares, tratamento de infecções recorrentes, suporte para problemas de coagulação e cuidados cardiológicos, quando necessário. Não há informações sobre cobertura de procedimentos pelo Sistema Único de Saúde (SUS) para essa condição.[1]
Tratamentos citados na literatura
Não há tratamentos específicos citados na literatura científica disponível nas fontes fornecidas. Portanto, esta seção não pode ser preenchida com base nos fatos autorizados.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Defeito no complexo de Golgi oligomérico conservado varia conforme a gravidade dos sintomas e o envolvimento de múltiplos órgãos. A deficiência intelectual grave e as infecções recorrentes podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e o suporte multidisciplinar são fundamentais para otimizar o bem-estar e a funcionalidade do paciente. Não há dados específicos sobre expectativa de vida nas fontes disponíveis.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
O complexo oligomérico de Golgi conservado, também conhecido como complexo COG, é uma proteína de membrana extrínseca encontrada no aparelho de Golgi em eucariotos. Este complexo é conservado evolutivamente e possui subunidades homólogas encontradas em todas as espécies de eucariotos. O complexo COG foi descoberto pela primeira vez em 1981 em células de ovário de hamster chinês. Essas células de ovário apresentavam mutações nos receptores de lipoproteína de baixa densidade (LDL), o que afetava a função das enzimas de glicosilação do Golgi. No entanto, o complexo COG como um todo não foi totalmente compreendido até 2004, quando uma deficiência em uma das subunidades do COG foi associada a distúrbios congênitos da glicosilação. Essas descobertas levaram à percepção de que o complexo COG desempenha um papel importante na glicosilação e na classificação de proteínas.
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
O Defeito no complexo de Golgi oligomérico conservado é uma doença genética rara que afeta o funcionamento de uma estrutura celular chamada complexo de Golgi. Esse complexo é responsável por modificar, empacotar e enviar proteínas para os lugares certos dentro da célula. Quando há um defeito nesse processo, diversos órgãos e sistemas do corpo podem ser afetados, resultando em uma ampla variedade de sintomas. A condição é causada por alterações (mutações) em genes que produzem as subunidades do complexo de Golgi oligomérico conservado (COG).[1][3]
Sinais e sintomas
Os sinais e sintomas do Defeito no complexo de Golgi oligomérico conservado podem variar muito de pessoa para pessoa, mas frequentemente incluem problemas neurológicos, dificuldades alimentares, infecções recorrentes e alterações na face e nos ossos. Entre os fenótipos (características observáveis) descritos na literatura estão: microcefalia secundária (cabeça pequena que se desenvolve após o nascimento), deficiência intelectual grave, atrofia do músculo esquelético, atrofia cortical temporal, agenesia do vermis cerebelar (parte do cerebelo não se forma), sequência de Pierre-Robin (que pode incluir queixo pequeno, língua para trás e fenda no palato), microtia (orelha pequena ou malformada), filtro labial profundo (sulco entre o nariz e o lábio superior mais marcado), borda do vermelhão espessa (lábios grossos), formato facial anormal, teto acetabular plano e fossa acetabular rasa (alterações no quadril), desvio ulnar do dedo (dedos tortos para o lado do cotovelo), hipertensão arterial pulmonar (pressão alta na artéria do pulmão), dilatação do septo atrial e defeito do septo atrial (problemas no coração), sepse neonatal (infecção generalizada no recém-nascido), infecções respiratórias e gastrointestinais recorrentes, diarreia intermitente, dificuldades alimentares, anormalidade da cascata de coagulação (problemas de coagulação do sangue), atividade reduzida da proteína S (um anticoagulante natural), hipoplasia do esmalte (esmalte dos dentes fino ou ausente) e pele de laranja (aspecto da pele com pequenas depressões).[1][3]
Causas genéticas
A doença é causada por mutações em genes que codificam as subunidades do complexo de Golgi oligomérico conservado (COG). Os genes já identificados como associados a essa condição são: COG1, COG2, COG4, COG5, COG6, COG7 e COG8. Cada um desses genes fornece instruções para a produção de uma peça (subunidade) do complexo COG. Mutações em qualquer um desses genes podem prejudicar a montagem ou a função do complexo, levando aos sintomas da doença. A herança (como a condição é passada de pais para filhos) não está especificada nas fontes disponíveis.[1][4]
Diagnóstico
O diagnóstico do Defeito no complexo de Golgi oligomérico conservado é baseado na avaliação clínica dos sinais e sintomas e confirmado por meio de testes genéticos. Existem 336 testes genéticos disponíveis para essa condição, e 311 variantes (alterações no DNA) já foram registradas no ClinVar, um banco de dados público que reúne informações sobre a relação entre variantes genéticas e doenças. O diagnóstico genético é essencial para diferenciar essa condição de outras doenças raras com sintomas semelhantes.[1][4]
Tratamento e manejo
Atualmente, não há um tratamento específico ou medicamento aprovado para o Defeito no complexo de Golgi oligomérico conservado. O manejo da doença é focado no alívio dos sintomas e no suporte às necessidades individuais de cada paciente. Isso pode incluir acompanhamento com equipe multidisciplinar (neurologista, geneticista, fisioterapeuta, fonoaudiólogo, nutricionista, entre outros), intervenções para dificuldades alimentares, tratamento de infecções recorrentes, suporte para problemas de coagulação e cuidados cardiológicos, quando necessário. Não há informações sobre cobertura de procedimentos pelo Sistema Único de Saúde (SUS) para essa condição.[1]
Tratamentos citados na literatura
Não há tratamentos específicos citados na literatura científica disponível nas fontes fornecidas. Portanto, esta seção não pode ser preenchida com base nos fatos autorizados.[1]
Prognóstico e qualidade de vida
O prognóstico para pessoas com Defeito no complexo de Golgi oligomérico conservado varia conforme a gravidade dos sintomas e o envolvimento de múltiplos órgãos. A deficiência intelectual grave e as infecções recorrentes podem impactar significativamente a qualidade de vida. O acompanhamento médico regular e o suporte multidisciplinar são fundamentais para otimizar o bem-estar e a funcionalidade do paciente. Não há dados específicos sobre expectativa de vida nas fontes disponíveis.[1][3]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 84 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 262 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
7 genes identificados com associação a esta condição.
Required for normal Golgi function
Cytoplasm, cytosolGolgi apparatus membrane
Congenital disorder of glycosylation 2I
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. Congenital disorder of glycosylation type 2I is characterized by mild neurological impairments.
Required for normal Golgi function (PubMed:19536132, PubMed:30290151). Plays a role in SNARE-pin assembly and Golgi-to-ER retrograde transport via its interaction with SCFD1 (PubMed:19536132)
Cytoplasm, cytosolGolgi apparatus membrane
Congenital disorder of glycosylation 2J
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions.
Required for normal Golgi function
Golgi apparatus membrane
Congenital disorder of glycosylation 2H
CDGs are a family of severe inherited diseases caused by a defect in protein N-glycosylation. They are characterized by under-glycosylated serum proteins. These multisystem disorders present with a wide variety of clinical features, such as disorders of the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions.
Required for normal Golgi function
Golgi apparatus membrane
Congenital disorder of glycosylation 2E
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions.
Required for normal Golgi function
Golgi apparatus membrane
Congenital disorder of glycosylation 2L
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. Clinical features of CDG2L include neonatal intractable focal seizures, vomiting, loss of consciousness, intracranial bleeding due to vitamin K deficiency, and death in infancy.
Required for normal Golgi morphology and function
Golgi apparatus membrane
Congenital disorder of glycosylation 2Q
A form of congenital disorder of glycosylation, a genetically heterogeneous group of autosomal recessive, multisystem disorders caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. The transmission pattern of CDG2Q is consistent with autosomal recessive inheritance.
Required for normal Golgi function
Golgi apparatus membrane
Congenital disorder of glycosylation 2G
A multisystem disorder caused by a defect in glycoprotein biosynthesis and characterized by under-glycosylated serum glycoproteins. Congenital disorders of glycosylation result in a wide variety of clinical features, such as defects in the nervous system development, psychomotor retardation, dysmorphic features, hypotonia, coagulation disorders, and immunodeficiency. The broad spectrum of features reflects the critical role of N-glycoproteins during embryonic development, differentiation, and maintenance of cell functions. Clinical features of CDG2G include failure to thrive, generalized hypotonia, growth retardation and mild psychomotor retardation. CDG2G is biochemically characterized by a defect in O-glycosylation as well as N-glycosylation.
Medicamentos aprovados (FDA)
1 medicamento encontrado nos registros da FDA americana.
Variantes genéticas (ClinVar)
311 variantes patogênicas registradas no ClinVar.
Vias biológicas (Reactome)
4 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Defeito no complexo de Golgi oligomérico conservado
Selecione um estado ou use sua localização para ver resultados.
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Pesquisa e ensaios clínicos
Nenhum ensaio clínico registrado para esta condição.
Publicações mais relevantes
COG5 deficiency disrupts cellular copper homeostasis and underlies the impaired mitochondrial OXPHOS function.
COG5, a subunit of the conserved oligomeric Golgi (COG) complex, plays a critical role in retrograde trafficking within the Golgi apparatus. Dysfunction of COG5 is associated with various human disorders, yet the underlying pathogenic mechanisms remain poorly understood. To investigate the mechanisms, we conducted proteomic analyses using COG5-deficient and rescue cell models, which revealed a potential link between COG5 dysfunction and mitochondrial oxidative phosphorylation (OXPHOS) deficiency. Using COG5-deficient cell models and patient-derived cells harboring COG5 variants, we biochemically validated the involvement of COG5 in mitochondrial OXPHOS, particularly in the regulation of complex I content. These models also exhibited elevated cellular copper levels. Notably, the significant reduction in OXPHOS complexes could be rescued by either restoring COG5 expression or administering a copper chelator. We further demonstrated that excessive cellular copper disrupts the function of mitochondrial iron-sulfur clusters, potentially leading to complex I assembly defects. Additionally, we identified a patient with biallelic COG5 variants presenting with a distinct subtype of mitochondrial disease (Leigh syndrome), a phenotype not previously associated with COG5-related disorders. These findings provide novel mechanistic insights into the role of COG5, extending beyond its established function in Golgi-mediated glycosylation modifications. Our results underscore the importance of COG5 in mitochondrial function through a copper-dependent pathway, offering new perspectives on its contribution to cellular homeostasis and disease pathogenesis.
The COG3 subunit interacts with SNX1 to enhance salt tolerance in Arabidopsis.
Salt stress severely restricts plant growth and agricultural productivity. The Conserved Oligomeric Golgi complex (COG) plays a key role in vesicular trafficking, but its function in plant stress responses remains poorly understood. Here, we demonstrated that the expression of the subunit COG3 of the COG complex in Arabidopsis thaliana was significantly induced under salt stress. The COG3 knockdown mutant (cog3) exhibits severe growth and development defects, as well as high sensitivity to salt stress. Furthermore, genetic complementation with COG3-GFP partially rescued the salt-sensitive phenotype of the cog3 mutant, while overexpression of COG3 enhanced plant salt tolerance. Mechanistically, COG3 physically interacts with the Sorting Nexin 1 (SNX1) on the endosome, and genetic evidence indicates that COG3 acts upstream of SNX1. We propose that under salt stress, COG3 maintains Golgi structural integrity and stabilizes SNX1 protein, thereby facilitating SNX1-mediated recycling of cargo proteins from endosomes to the plasma membrane to maintain cellular ion homeostasis. Loss of COG3 function disrupts this recycling pathway, leading to salt hypersensitivity. Our study reveals a novel COG3-SNX1 regulatory module that mediates protein recycling to enhance salt tolerance, providing new insights into the molecular mechanisms underlying plant adaptation to abiotic stress.
Identification of an ERGIC-Like Compartment in Fission Yeast: Emp43 Functions as a Lectin-Like Cargo Receptor for Glycosylated Proteins.
The endoplasmic reticulum-Golgi intermediate compartment (ERGIC) plays a crucial role in the secretory pathway; however, its existence and function in lower eukaryotes remain largely unexamined. In this study, we identified Emp43 (SPBC4F6.05c) of Schizosaccharomyces pombe, an orthologue of human (Homo sapiens) ERGIC-53, and demonstrated its localization to an ERGIC-like compartment. The localization of Emp43 depended on its C-terminal KYL motif and oligomerization through the CC1 domain. Deletion of S. pombe emp43+ resulted in significant sensitivity to MgCl2 and FK506, along with defects in septum integrity, indicating a role in cell wall maintenance. Further analysis identified Ssp120 of S. pombe, an orthologue of human MCFD2, as a functional partner of Emp43. Yeast two-hybrid assays confirmed a strong interaction between Emp43 and Ssp120, and both proteins co-localized within an ERGIC-like compartment. Additionally, we identified Meu17 of S. pombe, a glucan-α-1,4-glucosidase homolog, as a potential ligand for Emp43. Overexpression of Meu17 rescued MgCl2 sensitivity in both emp43Δ and ssp120Δ strains, while mutations in its N-linked glycosylation sites (N383, N409) or its predicted active site (D203) disrupted its septum localization and functional rescue capability. Our findings indicate that Emp43 forms a complex with Ssp120 to facilitate the transport of glycosylated proteins, such as Meu17, within an ERGIC-like compartment in fission yeast S. pombe. This study provides the first evidence of an ERGIC-like structure in S. pombe and highlights the conserved nature of ERGIC-associated mechanisms across eukaryotes.
Killer toxin K28 resistance in yeast relies on COG complex-mediated trafficking of the defence factor Ktd1.
AB toxins are a diverse family of protein toxins that enter host cells via endocytosis and induce cell death. In yeast, the AB toxin K28 is internalised to endosomes of susceptible yeast, before following the retrograde trafficking pathway and ultimately triggering cell cycle arrest. The endolysosomal defence factor Ktd1 protects against K28, but its regulation remains unclear. We show all lobe B subunits of the conserved oligomeric Golgi (COG) tethering complex are required for K28 resistance. Our experiments suggest the hypersensitivity of cog mutants is primarily explained by defects in Ktd1 trafficking. Ktd1 mis-localisation in cog mutants is reminiscent of disruptions in Snc1, a surface cargo that recycles multiple times via the Golgi. This work suggests not only that the COG complex is responsible for the precise trafficking of Ktd1 required to mediate toxin defence, but that Ktd1 might survey endolysosomal compartments for toxin. This work underpins the importance of Ktd1 in defence against the AB toxin K28, and implies how various membrane trafficking regulators could influence toxin effects in other eukaryotic systems.
Deep proteomic profiling of the intra-Golgi trafficking intermediates.
Intracellular trafficking relies on small membrane intermediates that transport cargo between different compartments. However, the precise role of vesicles in preserving Golgi function remains uncertain. To clarify this, we induced acute inactivation of the Conserved Oligomeric Golgi (COG) complex and analyzed vesicles from the different Golgi compartments. Proteomic analysis of the resulting vesicles revealed distinct molecular profiles, indicating a robust recycling system for Golgi proteins. All glycosylation enzymes and sugar transporters were detected in immunoisolated vesicles. The abundance of glycosylation machinery in intra-Golgi vesicles significantly increased following acute COG malfunction. Vesicles isolated from wild-type cells retained various vesicular coats, which were detaching from COG complex-dependent (CCD) vesicles stalled in the untethered state. Additionally, COG depletion led to increased molecular overlap among different populations of vesicles, suggesting that defects in vesicle tethering disrupt intra-Golgi sorting. Notably, CCD vesicles were functional and could be specifically rerouted to mitochondria that ectopically express Golgi tethers. Our findings demonstrate that the entire Golgi glycosylation machinery recycles within vesicles in a COG-dependent manner, whereas secretory and ER-Golgi trafficking proteins were not enriched. These results support a model in which the COG complex orchestrates the multistep recycling of glycosylation machinery, coordinated by specific coats, tethers, and SNAREs.
Publicações recentes
Mast cell mediators in hereditary angioedema.
Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
📚 EuropePMCmostrando 43
COG5 deficiency disrupts cellular copper homeostasis and underlies the impaired mitochondrial OXPHOS function.
PLoS geneticsThe COG3 subunit interacts with SNX1 to enhance salt tolerance in Arabidopsis.
Plant physiology and biochemistry : PPBIdentification of an ERGIC-Like Compartment in Fission Yeast: Emp43 Functions as a Lectin-Like Cargo Receptor for Glycosylated Proteins.
Molecular microbiologyKiller toxin K28 resistance in yeast relies on COG complex-mediated trafficking of the defence factor Ktd1.
Journal of cell scienceDeep proteomic profiling of the intra-Golgi trafficking intermediates.
Molecular biology of the cellComprehensive Proteomic Characterization of the Intra-Golgi Trafficking Intermediates.
bioRxiv : the preprint server for biologyCharacterization and functional analysis of Toxoplasma Golgi-associated proteins identified by proximity labeling.
mBioSyntaxin-5's flexibility in SNARE pairing supports Golgi functions.
Traffic (Copenhagen, Denmark)Rapid COG Depletion in Mammalian Cell by Auxin-Inducible Degradation System.
Methods in molecular biology (Clifton, N.J.)Acute COG complex inactivation unveiled its immediate impact on Golgi and illuminated the nature of intra-Golgi recycling vesicles.
Traffic (Copenhagen, Denmark)COG6-CDG: Novel variants and novel malformation.
Birth defects researchDevelopment and Initial Characterization of Cellular Models for COG Complex-Related CDG-II Diseases.
Frontiers in geneticsA Dominant Heterozygous Mutation in COG4 Causes Saul-Wilson Syndrome, a Primordial Dwarfism, and Disrupts Zebrafish Development via Wnt Signaling.
Frontiers in cell and developmental biologyGenetic analysis and prenatal diagnosis in a Chinese with growth retardation, abnormal liver function, and microcephaly.
Molecular genetics & genomic medicineProteoglycan synthesis in conserved oligomeric Golgi subunit deficient HEK293T cells is affected differently, depending on the lacking subunit.
Traffic (Copenhagen, Denmark)COG1-congenital disorders of glycosylation: Milder presentation and review.
Clinical geneticsSaul-Wilson Syndrome Missense Allele Does Not Show Obvious Golgi Defects in a C. elegans Model.
microPublication biologySyntaxin of plants31 (SYP31) and SYP32 is essential for Golgi morphology maintenance and pollen development.
Plant physiologyDisorder of sex development associated with a novel homozygous nonsense mutation in COG6 expands the phenotypic spectrum of COG6-CDG.
American journal of medical genetics. Part AGolgi inCOGnito: From vesicle tethering to human disease.
Biochimica et biophysica acta. General subjectsSugary Logistics Gone Wrong: Membrane Trafficking and Congenital Disorders of Glycosylation.
International journal of molecular sciencesArabidopsis COG6 is essential for pollen tube growth and Golgi structure maintenance.
Biochemical and biophysical research communicationsMaintaining order: COG complex controls Golgi trafficking, processing, and sorting.
FEBS lettersDefects in COG-Mediated Golgi Trafficking Alter Endo-Lysosomal System in Human Cells.
Frontiers in cell and developmental biologyA Recurrent De Novo Heterozygous COG4 Substitution Leads to Saul-Wilson Syndrome, Disrupted Vesicular Trafficking, and Altered Proteoglycan Glycosylation.
American journal of human geneticsDefective mucin-type glycosylation on α-dystroglycan in COG-deficient cells increases its susceptibility to bacterial proteases.
The Journal of biological chemistryDetailed Analysis of the Interaction of Yeast COG Complex.
Cell structure and functionMore than just sugars: Conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects.
Traffic (Copenhagen, Denmark)Conserved Oligomeric Golgi and Neuronal Vesicular Trafficking.
Handbook of experimental pharmacologyA Mild Form of COG5 Defect Showing Early-Childhood-Onset Friedreich's-Ataxia-Like Phenotypes with Isolated Cerebellar Atrophy.
Journal of Korean medical scienceCOG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes.
Journal of cell scienceHypothesis: lobe A (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG.
Journal of medical geneticsThe Golgin protein Coy1 functions in intra-Golgi retrograde transport and interacts with the COG complex and Golgi SNAREs.
Molecular biology of the cellThe Arl3 and Arl1 GTPases co-operate with Cog8 to regulate selective autophagy via Atg9 trafficking.
Traffic (Copenhagen, Denmark)MALDI-MS profiling of serum O-glycosylation and N-glycosylation in COG5-CDG.
Journal of mass spectrometry : JMSCreating Knockouts of Conserved Oligomeric Golgi Complex Subunits Using CRISPR-Mediated Gene Editing Paired with a Selection Strategy Based on Glycosylation Defects Associated with Impaired COG Complex Function.
Methods in molecular biology (Clifton, N.J.)The role of acroblast formation during Drosophila spermatogenesis.
Biology openArabidopsis COG Complex Subunits COG3 and COG8 Modulate Golgi Morphology, Vesicle Trafficking Homeostasis and Are Essential for Pollen Tube Growth.
PLoS geneticsCOG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe A sub-complex.
Scientific reportsGlobal serum glycoform profiling for the investigation of dystroglycanopathies & Congenital Disorders of Glycosylation.
Molecular genetics and metabolism reportsCOG Complex Complexities: Detailed Characterization of a Complete Set of HEK293T Cells Lacking Individual COG Subunits.
Frontiers in cell and developmental biologyDefects in the COG complex and COG-related trafficking regulators affect neuronal Golgi function.
Frontiers in neuroscienceMALDI-TOF MS applied to apoC-III glycoforms of patients with congenital disorders affecting O-glycosylation. Comparison with two-dimensional electrophoresis.
Proteomics. Clinical applicationsAssociações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Defeito no complexo de Golgi oligomérico conservado.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Defeito no complexo de Golgi oligomérico conservado
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- COG5 deficiency disrupts cellular copper homeostasis and underlies the impaired mitochondrial OXPHOS function.
- The COG3 subunit interacts with SNX1 to enhance salt tolerance in Arabidopsis.
- Identification of an ERGIC-Like Compartment in Fission Yeast: Emp43 Functions as a Lectin-Like Cargo Receptor for Glycosylated Proteins.
- Killer toxin K28 resistance in yeast relies on COG complex-mediated trafficking of the defence factor Ktd1.
- Deep proteomic profiling of the intra-Golgi trafficking intermediates.
- Mast cell mediators in hereditary angioedema.
- Prenatal Molecular Diagnosis of COL2A1-Associated Stickler Syndrome: Genotype-Phenotype Correlation in a Resource-Limited Healthcare Setting.
- Platelet gene signatures detecting pulmonary artery stenosis in patients with pulmonary hypertension.
- The global impact of imiglucerase therapy in children with Gaucher disease types 1 and 3: a real-world analysis from the International Collaborative Gaucher Group Gaucher Registry.
- Monogenic lupus with SLC7A7 mutations: a retrospective study from a Chinese center.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:309568(Orphanet)
- MONDO:0017750(MONDO)
- GARD:21344(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55787326(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
Defeito no complexo de Golgi oligomérico conservado
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata
- Medicamentos aprovados FDA
- fonte: FDA OpenFDA