Esta é uma lista de doenças que começam com a letra "S".
Introdução
O que você precisa saber de cara
Visão geral
A Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual é uma condição genética rara que afeta múltiplos sistemas do corpo. Ela é caracterizada por problemas progressivos de visão (incluindo retinite pigmentosa e catarata que aparece ainda na juventude), baixa estatura e dificuldades no desenvolvimento intelectual. Por ser uma síndrome muito rara, sua prevalência é estimada em menos de 1 caso para cada 1.000.000 de pessoas.[1][4]
Sinais e sintomas
Os sinais e sintomas geralmente começam na infância e incluem uma combinação de alterações oculares, de crescimento e neurológicas. Os principais sintomas oculares são: cegueira noturna progressiva (dificuldade para enxergar em ambientes com pouca luz), perda progressiva da visão, constrição do campo visual periférico (visão em túnel), catarata juvenil (opacificação do cristalino que surge precocemente), distrofia de cones e bastonetes (degeneração das células da retina) e atrofia em placas do epitélio pigmentar da retina. Exames como potenciais evocados visuais podem se tornar indetectáveis com a progressão da doença.[1][4]
Em relação ao crescimento e desenvolvimento, a síndrome causa baixa estatura, atraso global do desenvolvimento, deficiência intelectual de grau moderado, coordenação motora fina pobre e incoordenação motora geral. Também são observadas características físicas distintas, como: fissura palpebral ascendente ou inclinada para baixo, micrognatia (queixo pequeno), columela larga (parte do nariz entre as narinas), asas nasais subdesenvolvidas, achatamento malar (maçãs do rosto achatadas), macrotia (orelhas grandes), lobo da orelha aderido, orelhas de implantação baixa, pescoço curto, braquidactilia (dedos das mãos e pés curtos) e má oclusão dentária.[1][4]
Causas genéticas
Esta síndrome é causada por alterações (mutações) no gene RDH11 (Retinol dehydrogenase 11). Este gene fornece instruções para a produção de uma enzima importante para o ciclo da visão e para o metabolismo da vitamina A. Mutações neste gene interrompem esse processo, levando aos problemas oculares e aos outros sintomas da síndrome.[1][2][5]
A herança é autossômica recessiva, o que significa que a pessoa precisa herdar uma cópia do gene alterado do pai e outra da mãe para desenvolver a doença. Pais que são portadores de uma única cópia do gene alterado geralmente não apresentam sintomas.[1][2]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos e confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é um dos principais métodos para identificar mutações no gene RDH11. Outros exames genéticos que podem ser utilizados incluem o cariótipo (bandas G, Q ou R) e a pesquisa de microdeleções/microduplicações por FISH. A dosagem de alfa-fetoproteína também pode ser solicitada como parte da investigação.[1][2][5]
Atualmente, existem 19 variantes do gene RDH11 associadas a esta condição registradas no ClinVar, e 8 tipos de testes genéticos disponíveis para o diagnóstico. O código CID-10 para esta síndrome é Q87.8 (outras síndromes de anomalias congênitas especificadas).[1][2][5]
Tratamento e manejo
Não existe cura para a síndrome, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O acompanhamento deve ser multidisciplinar, envolvendo oftalmologistas, geneticistas, endocrinologistas, neurologistas, fisioterapeutas e terapeutas ocupacionais. Para a deficiência visual, podem ser indicados recursos de baixa visão e adaptações ambientais. A catarata juvenil pode necessitar de cirurgia para remoção do cristalino opaco. O atraso no desenvolvimento e a deficiência intelectual se beneficiam de estimulação precoce, terapia educacional e suporte psicossocial.[1][2]
No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo procedimentos como: cariótipo, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES), dosagem de alfa-fetoproteína e atendimento em reabilitação para doenças raras.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável, mas a condição geralmente leva a uma perda visual progressiva ao longo da vida, podendo resultar em cegueira legal. A deficiência intelectual moderada e a baixa estatura são permanentes. Com acompanhamento médico adequado, suporte educacional e terapias de reabilitação, é possível maximizar a independência e a qualidade de vida da pessoa afetada.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Esta é uma lista de doenças que começam com a letra "S".
Escala de raridade
<1/50kMuito rara
1/20kRara
1/10kPouco freq.
1/5kIncomum
1/2k
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Entender a doença
Do básico ao detalhe, leia no seu ritmo
Preparando trilha educativa...
Sinais e sintomas
O que aparece no corpo e com que frequência cada sintoma acontece
Visão geral
A Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual é uma condição genética rara que afeta múltiplos sistemas do corpo. Ela é caracterizada por problemas progressivos de visão (incluindo retinite pigmentosa e catarata que aparece ainda na juventude), baixa estatura e dificuldades no desenvolvimento intelectual. Por ser uma síndrome muito rara, sua prevalência é estimada em menos de 1 caso para cada 1.000.000 de pessoas.[1][4]
Sinais e sintomas
Os sinais e sintomas geralmente começam na infância e incluem uma combinação de alterações oculares, de crescimento e neurológicas. Os principais sintomas oculares são: cegueira noturna progressiva (dificuldade para enxergar em ambientes com pouca luz), perda progressiva da visão, constrição do campo visual periférico (visão em túnel), catarata juvenil (opacificação do cristalino que surge precocemente), distrofia de cones e bastonetes (degeneração das células da retina) e atrofia em placas do epitélio pigmentar da retina. Exames como potenciais evocados visuais podem se tornar indetectáveis com a progressão da doença.[1][4]
Em relação ao crescimento e desenvolvimento, a síndrome causa baixa estatura, atraso global do desenvolvimento, deficiência intelectual de grau moderado, coordenação motora fina pobre e incoordenação motora geral. Também são observadas características físicas distintas, como: fissura palpebral ascendente ou inclinada para baixo, micrognatia (queixo pequeno), columela larga (parte do nariz entre as narinas), asas nasais subdesenvolvidas, achatamento malar (maçãs do rosto achatadas), macrotia (orelhas grandes), lobo da orelha aderido, orelhas de implantação baixa, pescoço curto, braquidactilia (dedos das mãos e pés curtos) e má oclusão dentária.[1][4]
Causas genéticas
Esta síndrome é causada por alterações (mutações) no gene RDH11 (Retinol dehydrogenase 11). Este gene fornece instruções para a produção de uma enzima importante para o ciclo da visão e para o metabolismo da vitamina A. Mutações neste gene interrompem esse processo, levando aos problemas oculares e aos outros sintomas da síndrome.[1][2][5]
A herança é autossômica recessiva, o que significa que a pessoa precisa herdar uma cópia do gene alterado do pai e outra da mãe para desenvolver a doença. Pais que são portadores de uma única cópia do gene alterado geralmente não apresentam sintomas.[1][2]
Diagnóstico
O diagnóstico é baseado na avaliação clínica dos sintomas característicos e confirmado por testes genéticos. O sequenciamento completo do exoma (WES) é um dos principais métodos para identificar mutações no gene RDH11. Outros exames genéticos que podem ser utilizados incluem o cariótipo (bandas G, Q ou R) e a pesquisa de microdeleções/microduplicações por FISH. A dosagem de alfa-fetoproteína também pode ser solicitada como parte da investigação.[1][2][5]
Atualmente, existem 19 variantes do gene RDH11 associadas a esta condição registradas no ClinVar, e 8 tipos de testes genéticos disponíveis para o diagnóstico. O código CID-10 para esta síndrome é Q87.8 (outras síndromes de anomalias congênitas especificadas).[1][2][5]
Tratamento e manejo
Não existe cura para a síndrome, e o tratamento é focado no manejo dos sintomas e na melhora da qualidade de vida. O acompanhamento deve ser multidisciplinar, envolvendo oftalmologistas, geneticistas, endocrinologistas, neurologistas, fisioterapeutas e terapeutas ocupacionais. Para a deficiência visual, podem ser indicados recursos de baixa visão e adaptações ambientais. A catarata juvenil pode necessitar de cirurgia para remoção do cristalino opaco. O atraso no desenvolvimento e a deficiência intelectual se beneficiam de estimulação precoce, terapia educacional e suporte psicossocial.[1][2]
No Brasil, o Sistema Único de Saúde (SUS) oferece cobertura mínima para esta condição, incluindo procedimentos como: cariótipo, pesquisa de microdeleções/microduplicações por FISH, sequenciamento completo do exoma (WES), dosagem de alfa-fetoproteína e atendimento em reabilitação para doenças raras.[1][2]
Prognóstico e qualidade de vida
O prognóstico é variável, mas a condição geralmente leva a uma perda visual progressiva ao longo da vida, podendo resultar em cegueira legal. A deficiência intelectual moderada e a baixa estatura são permanentes. Com acompanhamento médico adequado, suporte educacional e terapias de reabilitação, é possível maximizar a independência e a qualidade de vida da pessoa afetada.[1][4]
Conteúdo informativo gerado e mantido automaticamente a partir de fontes oficiais (Orphanet, HPO, OMIM, SUS). Não substitui avaliação médica.
Partes do corpo afetadas
+ 11 sintomas em outras categorias
Características mais comuns
Os sintomas variam de pessoa para pessoa. Abaixo estão as 36 características clínicas mais associadas, ordenadas por frequência.
Linha do tempo da pesquisa
Encontrou um erro ou informação desatualizada? Sugira uma correção →
Genética e causas
O que está alterado no DNA e como passa nas famílias
Genes associados
1 gene identificado com associação a esta condição. Padrão de herança: Autosomal recessive.
Curadoria gene-doença
fontes oficiaisRetinol dehydrogenase with a clear preference for NADP. Displays high activity towards 9-cis, 11-cis and all-trans-retinol, and to a lesser extent on 13-cis-retinol (PubMed:12036956, PubMed:12226107, PubMed:29410696). Exhibits a low reductive activity towards unsaturated medium-chain aldehydes such as cis -6-nonenal and no activity toward nonanal or 4-hydroxy-nonenal (PubMed:15865448). Has no dehydrogenase activity towards steroid (PubMed:12036956, PubMed:12226107)
Endoplasmic reticulum membrane
Retinal dystrophy, juvenile cataracts, and short stature syndrome
A disorder characterized by retinal dystrophy resulting in progressive decrease in visual acuity and difficulties with night vision in the first decade of life, development of juvenile cataracts, facial dysmorphism, psychomotor developmental delays, learning disabilities and short stature. Ophthalmological findings include salt-and-pepper retinopathy, attenuation of the arterioles, generalized rod-cone dysfunction, mottled macula at an early age, and peripapillary sparing of the retinal pigment epithelium.
Variantes genéticas (ClinVar)
19 variantes patogênicas registradas no ClinVar.
Classificação de variantes (ClinVar)
Distribuição de 5 variantes classificadas pelo ClinVar.
Vias biológicas (Reactome)
3 vias biológicas associadas aos genes desta condição.
Diagnóstico
Os sinais que médicos procuram e os exames que confirmam
Tratamento e manejo
Remédios, cuidados de apoio e o que precisa acompanhar
Onde tratar no SUS
Hospitais de referência no Brasil e o protocolo oficial do SUS (PCDT)
🇧🇷 Atendimento SUS — Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual
Centros de Referência SUS
13 centros habilitados pelo SUS para Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual
Centros para Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual
Detalhes dos centros
Hospital Infantil Albert Sabin
R. Tertuliano Sales, 544 - Vila União, Fortaleza - CE, 60410-794 · CNES 2407876
Serviço de Referência
Hospital de Apoio de Brasília (HAB)
AENW 3 Lote A Setor Noroeste - Plano Piloto, Brasília - DF, 70684-831 · CNES 0010456
Serviço de Referência
Hospital Estadual Infantil e Maternidade Alzir Bernardino Alves (HIABA)
Av. Min. Salgado Filho, 918 - Soteco, Vila Velha - ES, 29106-010 · CNES 6631207
Serviço de Referência
Hospital das Clínicas da UFMG
Av. Prof. Alfredo Balena, 110 - Santa Efigênia, Belo Horizonte - MG, 30130-100 · CNES 2280167
Serviço de Referência
Hospital Universitário João de Barros Barreto
R. dos Mundurucus, 4487 - Guamá, Belém - PA, 66073-000 · CNES 2337878
Serviço de Referência
Instituto de Medicina Integral Prof. Fernando Figueira (IMIP)
R. dos Coelhos, 300 - Boa Vista, Recife - PE, 50070-902 · CNES 0000647
Serviço de Referência
Hospital Pequeno Príncipe
R. Des. Motta, 1070 - Água Verde, Curitiba - PR, 80250-060 · CNES 3143805
Serviço de Referência
Hospital de Clínicas da UFPR
R. Gen. Carneiro, 181 - Alto da Glória, Curitiba - PR, 80060-900 · CNES 2364980
Serviço de Referência
Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira (IFF/Fiocruz)
Av. Rui Barbosa, 716 - Flamengo, Rio de Janeiro - RJ, 22250-020 · CNES 2269988
Serviço de Referência
Hospital de Clínicas de Porto Alegre (HCPA)
Rua Ramiro Barcelos, 2350 Bloco A - Av. Protásio Alves, 211 - Bloco B e C - Santa Cecília, Porto Alegre - RS, 90035-903 · CNES 2237601
Serviço de Referência
Hospital das Clínicas da FMUSP
R. Dr. Ovídio Pires de Campos, 225 - Cerqueira César, São Paulo - SP, 05403-010 · CNES 2077485
Serviço de Referência
Hospital de Clínicas da UNICAMP
R. Vital Brasil, 251 - Cidade Universitária, Campinas - SP, 13083-888 · CNES 2748223
Serviço de Referência
Hospital de Clínicas de Ribeirão Preto (HCRP-USP)
R. Ten. Catão Roxo, 3900 - Vila Monte Alegre, Ribeirão Preto - SP, 14015-010 · CNES 2082187
Serviço de Referência
Dados de DATASUS/CNES, SBGM, ABNeuro e Ministério da Saúde. Sempre confirme a disponibilidade diretamente com o estabelecimento.
Pesquisa ativa
Ensaios clínicos abertos e novidades científicas recentes
Ensaios em destaque
🟢 Recrutando agora
1 pesquisa recrutando participantes. Converse com seu médico sobre a possibilidade de participar.
Outros ensaios clínicos
Publicações mais relevantes
Publicações recentes
Novel variant causing OTUD6B-related syndrome with ocular dysplasia and hypothyroidism: the first Chinese case.
Poretti-Boltshauser Syndrome: A Potential Pathognomonic "Wolfjaw" Pattern of Retinal Perfusion.
DHX16-Associated Neuromuscular Oculoauditory Syndrome: A Novel Case.
Denovo variants in POGZ and YY1 genes: The novel mega players for neurodevelopmental syndromes in two unrelated consanguineous families.
A novel large multi-gene deletion in syndromic choroideremia.
Associações
Organizações que acompanham esta doença — pra ter apoio e orientação
Ainda não temos associações cadastradas para Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual.
É de uma associação que acompanha esta doença? Fale com a gente →
Comunidades
Grupos ativos de quem convive com esta doença aqui no Raras
Ainda não existe comunidade no Raras para Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual
Pacientes, familiares e cuidadores se organizam em comunidades pra compartilhar experiências, fazer perguntas e se apoiar. Você pode ser o primeiro.
Tire suas dúvidas
Perguntas, dicas e experiências compartilhadas aqui na página
Participe da discussão
Faça login para postar dúvidas, compartilhar experiências e interagir com especialistas.
Fazer loginDoenças relacionadas
Doenças com sintomas parecidos — ajudam quem ainda está buscando diagnóstico
Referências e fontes
Bases de dados externas citadas neste artigo
Publicações científicas
Artigos indexados no PubMed ligados a esta doença no grafo RarasNet — título, periódico e PMID direto da fonte, sem intermediação de IA.
- Novel variant causing OTUD6B-related syndrome with ocular dysplasia and hypothyroidism: the first Chinese case.
- Poretti-Boltshauser Syndrome: A Potential Pathognomonic "Wolfjaw" Pattern of Retinal Perfusion.
- DHX16-Associated Neuromuscular Oculoauditory Syndrome: A Novel Case.
- Denovo variants in POGZ and YY1 genes: The novel mega players for neurodevelopmental syndromes in two unrelated consanguineous families.
- A novel large multi-gene deletion in syndromic choroideremia.
Bases de dados e fontes oficiais
Identificadores e referências canônicas usadas para montar este verbete.
- ORPHA:436245(Orphanet)
- OMIM OMIM:616108(OMIM)
- MONDO:0014495(MONDO)
- GARD:17730(GARD (NIH))
- Variantes catalogadas(ClinVar)
- Busca completa no PubMed(PubMed)
- Q55784851(Wikidata)
Dados compilados pelo RarasNet a partir de fontes abertas (Orphanet, OMIM, MONDO, PubMed/EuropePMC, ClinicalTrials.gov, DATASUS, PCDT/MS). Este conteúdo é informativo e não substitui avaliação médica.
Conteúdo mantido por Agente Raras · Médicos e pesquisadores podem colaborar
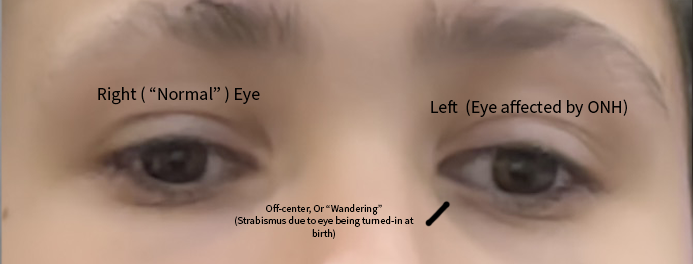
Síndrome de retinite pigmentosa-catarata juvenil-baixa estatura-perturbação do desenvolvimento intelectual
📋 Origem dos dados
Esta página agrega dados de fontes públicas e oficiais. Dados sobre cobertura no SUS (PCDT, CEAF) são verificados ativamente por agente proativo (ver badge no infobox). Demais dados têm atribuição de fonte + data da última sincronização — clique para abrir o original.
- Doença rara (ontologia)
- fonte: Orphanet
- Identificador unificado
- fonte: MONDO
- Genética mendeliana
- fonte: OMIM
- Codificação WHO/SUS
- fonte: WHO ICD-10 / DATASUS
- CID-11 (futuro)
- fonte: WHO ICD-11
- NIH/GARD
- fonte: GARD (NIH)
- Dado público estruturado
- fonte: Wikidata